
Advanced Nanocatalysis Lab
School of Engergy and Chemical Engineering
Publications
Filter by type:
123. Mild Hydrolysis of PET and Electrochemical Energy Recovery via Multifunctional Polyoxometalate Catalysts
Journal paper122. Diverse Cation Exchange in Layered Titanate Nanostructures for Tailored Catalysis
Journal paperLayered titanates (LTs) offer exceptional structural and chemical tunability, enabling precise modulation of their electronic states and catalytic properties. However, systematic studies on the range of cations that can serve as intercalants for LTs remain limited, and conventional synthesis methods often require additional treatments for cation intercaltion. In this study, we present a cation-free H+ (H3O+)-intercalated LT as a versatile platform for direct cation insertion. This LT can be intercalated with single metal cations (42 metals from five groups) or a combination of 5–30 cations without structural deformation. Intercalation with alkali metals (AMs) precisely tuned the charge density of Rh species when the prepared LTs are used as catalytic supports. Among the Rh-loaded AM-bearing LTs, Rh/K–LT delivered the highest turnover frequency (23 685 h−1), surpassing those of other AM-intercalated systems and previously reported Rh-based heterogeneous catalysts, during propylene hydroformylation. Combined in situ/ex situ analyses and density functional theory calculations revealed that AM intercalation promotes charge transfer to Rh, thereby enhancing adsorption behavior and catalytic activity. This work establishes not only a broad cation intercalation library but also a generalizable strategy for cation engineering in LTs, highlighting the potential of intercalation-driven charge modulation for rational catalyst design across diverse reactions.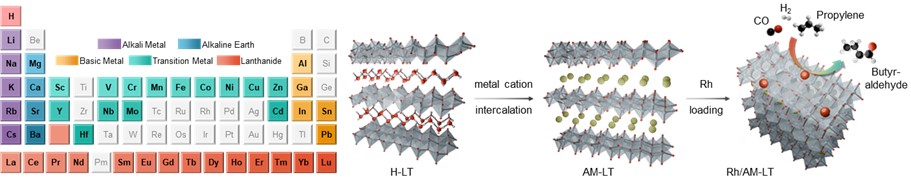
121. Rh cluster catalysts with enhanced catalytic activity: The 'Goldilocks Rh size' for olefin hydroformylation
Journal paperThis study investigated the performances of Rh single-atom catalysts (SACs), cluster catalysts, and nanoparticle (NP) catalysts in olefin hydroformylation. Using in situ characterization techniques, we elucidated the distinct chemical and electronic properties of each catalyst type. Our findings revealed that Rh cluster catalysts exhibit distinctive characteristics between those of SAC and NPs, significantly influencing their catalytic performance. Notably, Rh cluster catalysts a chieved a five-fold increase in turnover frequency (~25,589 h−1) compared to SACs (~5,430 h−1) and a nine-fold increase relative to NP catalysts (~2,838 h−1) in the propylene hydroformylation. Theoretical calculations revealed that the Rh cluster catalysts possess optimal CO adsorption energies, allowing them to efficiently overcome the energy barrier for CO insertion during the rate-determining step of propylene hydroformylation. Additionally, density of states and crystalline orbital Hamilton population analyses confirmed that the Rh cluster catalyst exhibited adjusted electronic properties, positioned between those of Rh SAC and NP catalysts. This study highlights the unique properties of the Rh cluster catalysts and offers valuable insights into the design of high-performance catalysts for hydroformylation and other catalytic processes.
chieved a five-fold increase in turnover frequency (~25,589 h−1) compared to SACs (~5,430 h−1) and a nine-fold increase relative to NP catalysts (~2,838 h−1) in the propylene hydroformylation. Theoretical calculations revealed that the Rh cluster catalysts possess optimal CO adsorption energies, allowing them to efficiently overcome the energy barrier for CO insertion during the rate-determining step of propylene hydroformylation. Additionally, density of states and crystalline orbital Hamilton population analyses confirmed that the Rh cluster catalyst exhibited adjusted electronic properties, positioned between those of Rh SAC and NP catalysts. This study highlights the unique properties of the Rh cluster catalysts and offers valuable insights into the design of high-performance catalysts for hydroformylation and other catalytic processes.

120. Upcycling waste polystyrene to adipic acid through a hybrid chemical and biological process
Journal paperOxidative catalytic depolymerization of polystyrene (PS) can produce benzoic acid, but the annual consumption of benzoic acid is ~40 times lower than PS. For this catalytic oxidation method to be a viable means to manage PS waste, benzoic acid should be converted to higher-volume chemicals. We demonstrate a hyb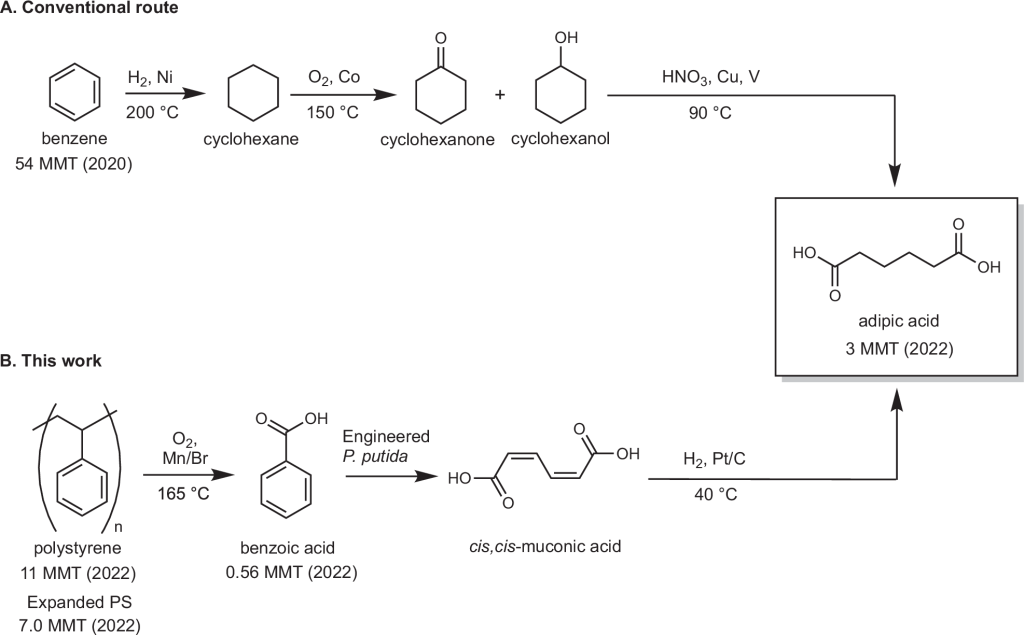 rid chemical and biological process that uses PS as feedstock for production of adipic acid, a high-volume co-monomer for nylon 6,6 via benzoic acid. Mn/Br co-catalyzed autoxidation of PS to benzoic acid proceeds with a yield of up to 94% in a solvent mixture of benzoic acid and water. The PS-derived benzoic acid undergoes bioconversion at near-quantitative yield to muconic acid, which is readily converted to adipic acid through catalytic hydrogenation. Process modeling, techno-economic analysis, and life cycle assessment estimate an adipic acid minimum selling price of $3.18/kg, with a 61% decrease in greenhouse gas emissions relative to production from fossil fuels.
rid chemical and biological process that uses PS as feedstock for production of adipic acid, a high-volume co-monomer for nylon 6,6 via benzoic acid. Mn/Br co-catalyzed autoxidation of PS to benzoic acid proceeds with a yield of up to 94% in a solvent mixture of benzoic acid and water. The PS-derived benzoic acid undergoes bioconversion at near-quantitative yield to muconic acid, which is readily converted to adipic acid through catalytic hydrogenation. Process modeling, techno-economic analysis, and life cycle assessment estimate an adipic acid minimum selling price of $3.18/kg, with a 61% decrease in greenhouse gas emissions relative to production from fossil fuels.
119. Ligand-Substrate Self-Sorting in Silica-Encased Rh-Nanopetals as Model Catalyst for Hydroformylation of Arylalkenes
Journal paperSelf-regulated ligand–substrate interaction with the metal center is fundamental to enzymes and homogeneous complexes, yet similar phenomena in heterogeneous systems remain underexplored. Simultaneous control of reactivity and selectivity becomes challenging due to the competitive molecular-adsorp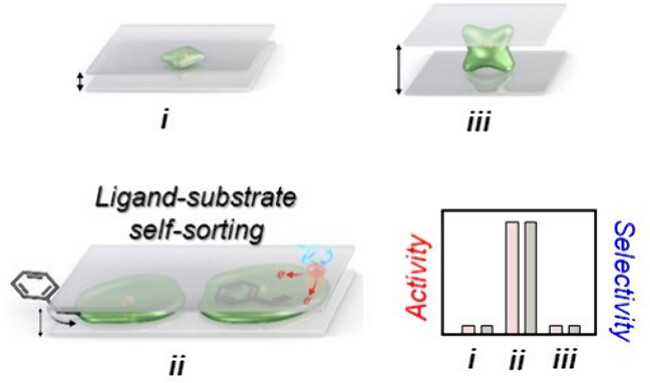 tion-mediated blocking of reaction-sites. Here, we address this dichotomy by introducing the concept of ligand–substrate self-sorting (LSS) in a silica-encased Rh-nanopetals (Rh-NPLs) model platform for hydroformylation of alkene. We synthesized different stages of Rh-NPLs (bud, bloomed flower and flower-in-book) inside a bilayer silica-encase. 2D-constrained Rh growth establishes controllable intimacy with the silica overlayer while availing molecular size interfacial spaces. Optimal design endows a well-orchestrated microenvironment to avoid conflicting ligand–substrate adsorption. Remarkably, LSS phenomena lead to high hydroformylation regioselectivity (>90%) without compromising the catalyst activity (>99% yield). The present work demonstrates the nanoscale material design capability, influencing the multimolecular dynamic events with implications toward sustainable chemical synthesis.
tion-mediated blocking of reaction-sites. Here, we address this dichotomy by introducing the concept of ligand–substrate self-sorting (LSS) in a silica-encased Rh-nanopetals (Rh-NPLs) model platform for hydroformylation of alkene. We synthesized different stages of Rh-NPLs (bud, bloomed flower and flower-in-book) inside a bilayer silica-encase. 2D-constrained Rh growth establishes controllable intimacy with the silica overlayer while availing molecular size interfacial spaces. Optimal design endows a well-orchestrated microenvironment to avoid conflicting ligand–substrate adsorption. Remarkably, LSS phenomena lead to high hydroformylation regioselectivity (>90%) without compromising the catalyst activity (>99% yield). The present work demonstrates the nanoscale material design capability, influencing the multimolecular dynamic events with implications toward sustainable chemical synthesis.
118. Hydrogen-Free Hydrodeoxygenation of Guaiacol using Phase Controlled NiAl Layered Double Hydroxides-derived Catalysts
Journal paperThe effective and sustainable conversion of lignin-derived aromatics into valuable chemicals is pivotal in reducing dependence on fossil-based resources. This study investigates the selective hydrodeoxygenation (HDO) of guaiacol via aqueous-phase reforming of methanol in the absence of external hydrogen, targeting the selective cleavage of the methoxy group in guaiacol and the hydrogenation of the aromatic ring. Highly dispersed Ni catalysts were prepared through nanoscale phase transformation of NiAl-layered double hydroxide (NiAl LDH) precursors. We demonstrate that increasing calcination temperatures enhanc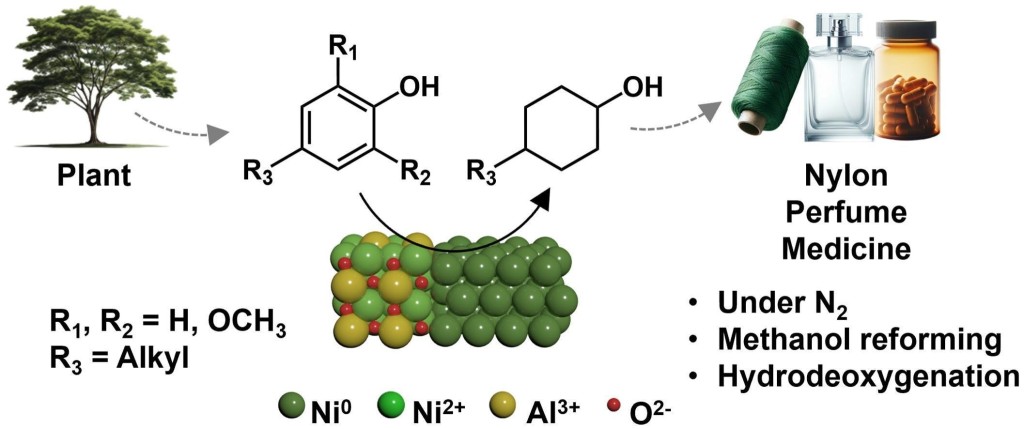 es the surface concentration of Ni0 nanophases while decreasing Ni2+ content. While metallic Ni serves as a key active site, the controlled NiAlOx phases, functioning as acid sites, are essential for stabilizing NiO and boosting guaiacol conversion. The NiAl-500 catalyst exhibited higher performance in hydrogen-free conversion of guaiacol to cyclohexanol, outperforming many non-noble catalysts. Furthermore, the developed catalyst also converted other lignin-derived phenolics to alkyl cyclohexanols via selective demethoxylation and hydrogenation. In situ characterizations elucidated the guaiacol HDO mechanism, highlighting the synergistic roles of Ni2+ and Ni0, alongside a distinctive Ni–O–Al bond in the LDH-derived catalyst. This work paves the way for the development of highly efficient, hydrogen-free HDO catalysts with high Ni dispersibility and content (>70 wt%) through precise tuning of the physical and chemical properties of LDH-based systems.
es the surface concentration of Ni0 nanophases while decreasing Ni2+ content. While metallic Ni serves as a key active site, the controlled NiAlOx phases, functioning as acid sites, are essential for stabilizing NiO and boosting guaiacol conversion. The NiAl-500 catalyst exhibited higher performance in hydrogen-free conversion of guaiacol to cyclohexanol, outperforming many non-noble catalysts. Furthermore, the developed catalyst also converted other lignin-derived phenolics to alkyl cyclohexanols via selective demethoxylation and hydrogenation. In situ characterizations elucidated the guaiacol HDO mechanism, highlighting the synergistic roles of Ni2+ and Ni0, alongside a distinctive Ni–O–Al bond in the LDH-derived catalyst. This work paves the way for the development of highly efficient, hydrogen-free HDO catalysts with high Ni dispersibility and content (>70 wt%) through precise tuning of the physical and chemical properties of LDH-based systems.
117. Upcycling Post-Consumer Polystyrene Waste into Liquid Organic Hydrogen Carriers
Journal paperTo address the dual challenges of plastic waste management and sustainable hydrogen storage, we propose an innovative strategy to upcycle post-consumer polystyrene (PS) waste into liquid organic hydrogen carriers (LOHCs). The process involves pyrolyzing PS into aromatic monomers, primarily styrene, which are subsequently hydrogenated into cyclic hydrocarbons such as ethylcyclohexane. Dehydrogenation of these hydrogen-rich LOHCs releases high-purity hydrogen, with catalytic performance strongly influenced by the properties of supported Pt catalysts. Among various supports, Pt catalysts on nanosheet-assembled Al2O3 demonstrated superior activity and stability, attributed to a higher proportion of metallic Pt0 species, low surface acidity, and enhanced pore structures. However, polycyclic compounds in the PS-derived LOHCs led to catalyst deactivation via coke formation, necessitating a distillation step to remove these precursors. Integration of distillation, energy recovery, and LOHC recycling were shown to maintain catalyst longevity and process efficiency. Life cycle assessment and techno-economic analysis confirmed that upcycling PS waste into LOHCs offers both environmental benefits, including a negative carbon footprint for LOHC production, and economic viability, with competitive hydrogen transport costs. This work not only presents a feasible route for converting PS waste into valuable energy carriers but also contributes to advancing circular carbon strategies and the hydrogen economy.
and process efficiency. Life cycle assessment and techno-economic analysis confirmed that upcycling PS waste into LOHCs offers both environmental benefits, including a negative carbon footprint for LOHC production, and economic viability, with competitive hydrogen transport costs. This work not only presents a feasible route for converting PS waste into valuable energy carriers but also contributes to advancing circular carbon strategies and the hydrogen economy.

116. Unveiling the role of Metal–Support interactions in Ni catalysts for CO2-Free hydrogen and carbon nanotube production via methane pyrolysis
Journal paperAs the demand for hydrogen (H2) production rises alongside the global push for net-zero CO2 emissions, methane pyrolysis (MP) has emerged as a promising technology for CO2-free H2 generation. Ni-based catalysts are widely studied for MP due to their ability to activate CH4 in the low reaction temperature, but rapid deactivation caused by carbon by-products remains a challenge. In this study, we investigated the role of metal-support interactions on the catalytic performance and carbon nanotube (CNT) production in MP. A series of NiO-supported alumina catalysts were synthesized via a on e-step solution combustion synthesis method with controllable NiO nanoparticle (NP) sizes and loading levels. The optimized catalyst exhibited an activation energy of 54.2 kJ/mol and remained active for up to 9 h. All catalysts in the series generated high quality CNTs as byproducts of MP, exhibiting controllable diameters and varying degrees of graphitization. Comprehensive analysis indicated no direct correlation between carbon yield and structural properties of catalysts such as NP size, surface area, dispersion or oxidation states. In contrast, hydrogen temperature-programmed reduction measurements revealed that the carbon yield was closely linked to the presence of β2-type NiO species associated with an Al-rich mixed oxide phase. Catalysts with higher amounts of β2-type NiO showed increased carbon production, likely driven by improved carbon diffusion at the Ni–Al2O3 interface. These results emphasize the importance of fine-tuning metal-support interactions to achieve optimal catalytic performance, enabling efficient H2 and CNT generation simultaneously during MP.
e-step solution combustion synthesis method with controllable NiO nanoparticle (NP) sizes and loading levels. The optimized catalyst exhibited an activation energy of 54.2 kJ/mol and remained active for up to 9 h. All catalysts in the series generated high quality CNTs as byproducts of MP, exhibiting controllable diameters and varying degrees of graphitization. Comprehensive analysis indicated no direct correlation between carbon yield and structural properties of catalysts such as NP size, surface area, dispersion or oxidation states. In contrast, hydrogen temperature-programmed reduction measurements revealed that the carbon yield was closely linked to the presence of β2-type NiO species associated with an Al-rich mixed oxide phase. Catalysts with higher amounts of β2-type NiO showed increased carbon production, likely driven by improved carbon diffusion at the Ni–Al2O3 interface. These results emphasize the importance of fine-tuning metal-support interactions to achieve optimal catalytic performance, enabling efficient H2 and CNT generation simultaneously during MP.
115. Strategies for oxygen vacancy formation in CeO2-based materials for thermal catalysis
Journal paperCeO2 is a prominent support material for heterogeneous catalysis owing to its exceptional oxygen storage capa city. The CeO2 oxygen vacancy (VO) density critically influences thermal catalytic processes involving oxygen species, such as CO oxidation, CO2 hydrogenation, and volatile organic compound oxidation. This review examines recent strategies for controlling VO in CeO2, including lattice doping, nanostructure control, and defect engineering via external reduction, as well as their effects on thermal catalytic reactions. We present diverse in situ characterization techniques to elucidate the relationship between the lattice oxygen mobility and catalytic reactivity during reactions. Strategies combining multiple approaches to achieve synergistic CeO2 reducibility enhancement are discussed. A comprehensive exploration of VO regulation strategies provides insights for optimizing CeO2-based systems in oxygen-mediated thermal catalysis.
city. The CeO2 oxygen vacancy (VO) density critically influences thermal catalytic processes involving oxygen species, such as CO oxidation, CO2 hydrogenation, and volatile organic compound oxidation. This review examines recent strategies for controlling VO in CeO2, including lattice doping, nanostructure control, and defect engineering via external reduction, as well as their effects on thermal catalytic reactions. We present diverse in situ characterization techniques to elucidate the relationship between the lattice oxygen mobility and catalytic reactivity during reactions. Strategies combining multiple approaches to achieve synergistic CeO2 reducibility enhancement are discussed. A comprehensive exploration of VO regulation strategies provides insights for optimizing CeO2-based systems in oxygen-mediated thermal catalysis.
114. Effective Production of Liquid/Wax Fuels from Polyethylene Plastics Using Ru/Al2O3 Catalysts
Journal paperHydrogenolysis provides a promising pathway for converting polyolefin plastics into valuable liquid and wax fuels. This process involves dehydrogenation, C–C bond cleavage, and hydrogenation at the active metal sites of the catalyst. Controlling the nature of these metal sites is crucial to optimize overall reaction activity. In this study, Ru catalysts supported on nanosheet-assembled Al2O3 (NA-Al2O3) were used for the hydrogenolysis of polyethylene (PE). Unlike conventional Al2O3, NA-Al2O3 promotes Ru–Al bond formation, leading to stronger metal–support interactions. Under identical Ru loadings, these enhanced in teractions resulted in higher Ru dispersion and smaller Ru species on the NA-Al2O3 surface. To investigate the effect of Ru loading, a series of catalysts (xRu/NA-Al2O3, x = 0.5, 1, 5, and 8 wt% Ru) was synthesized, revealing that Ru particle size and electronic properties varied with Ru loading. Among them, the 1Ru/NA-Al2O3 catalyst, featuring optimally sized Ru species (~0.8 nm) and a tailored electronic structure, demonstrated the highest efficiency in PE hydrogenolysis by effectively suppressing successive C–C bond cleavage. This catalyst achieved an outstanding PE conversion rate of 1.15 x 103 gconverted PE·gRu–1·h–1 and a liquid/wax production rate of 9.23 x 102 gliquid/wax·gRu–1·h–1, highlighting its superior performance in catalytic PE hydrogenolysis.
teractions resulted in higher Ru dispersion and smaller Ru species on the NA-Al2O3 surface. To investigate the effect of Ru loading, a series of catalysts (xRu/NA-Al2O3, x = 0.5, 1, 5, and 8 wt% Ru) was synthesized, revealing that Ru particle size and electronic properties varied with Ru loading. Among them, the 1Ru/NA-Al2O3 catalyst, featuring optimally sized Ru species (~0.8 nm) and a tailored electronic structure, demonstrated the highest efficiency in PE hydrogenolysis by effectively suppressing successive C–C bond cleavage. This catalyst achieved an outstanding PE conversion rate of 1.15 x 103 gconverted PE·gRu–1·h–1 and a liquid/wax production rate of 9.23 x 102 gliquid/wax·gRu–1·h–1, highlighting its superior performance in catalytic PE hydrogenolysis.
113. Design and Application of Mesoporous Catalysts for Liquid-Phase Furfural Hydrogenation
Journal paperFurfural (FAL), a platform molecule derived from biomass through acid-catalyzed processes, holds significant potential for producing various value-added chemicals. Its unique chemical structure, comprising a furan ring and an aldehyde functional group, enables diverse transformation pathways to yield products such as furfuryl alcohol, furan, tetrahydrofuran, and other industrially relevant compounds. Consequently, optimizing catalytic processes for FAL conversion has garnered substantial attention, particularly in selectivity and efficiency. The liquid-phase hydrogenation of FAL has demonstrated advantages, including enhanced catalyst stability and higher product yields. Among the catalysts investigated, mesoporous materials have emerged as promising candidates because of their high surface area, tunable pore structure, and ability to support highly dispersed active sites. These attributes are critical for maximizing the catalytic performance across various reactions, including FAL hydrogenation. This review provides a comprehensive overview of recent advances in mesoporous catalyst design for FAL hydrogenation, focusing on synthesis strategies, metal dispersion control, and structural optimization to enhance catalytic perf ormance. It explores noble metal-based catalysts, particularly highly dispersed Pd systems, as well as transition-metal-based alternatives such as Co-, Cu-, and Ni-based mesoporous catalysts, highlighting their electronic structure, bimetallic interactions, and active site properties. Additionally, metal–organic frameworks are introduced as both catalysts and precursors for thermally derived materials. Finally, key challenges that require further investigation are discussed, including catalyst stability, deactivation mechanisms, strategies to reduce reliance on external hydrogen sources, and the impact of solvent effects on product selectivity. By integrating these insights, this review provides a comprehensive perspective on the development of efficient and sustainable catalytic systems for biomass valorization.
ormance. It explores noble metal-based catalysts, particularly highly dispersed Pd systems, as well as transition-metal-based alternatives such as Co-, Cu-, and Ni-based mesoporous catalysts, highlighting their electronic structure, bimetallic interactions, and active site properties. Additionally, metal–organic frameworks are introduced as both catalysts and precursors for thermally derived materials. Finally, key challenges that require further investigation are discussed, including catalyst stability, deactivation mechanisms, strategies to reduce reliance on external hydrogen sources, and the impact of solvent effects on product selectivity. By integrating these insights, this review provides a comprehensive perspective on the development of efficient and sustainable catalytic systems for biomass valorization.
112. Pd-catalyzed dehydrogenation enhanced by charge transfer from MoOx promoter
Journal paperIncorporating metal oxides is a sensible strategy for enhancing the efficiency of precious metals. Upon introducing molybdenum oxide, diverse coordination structures were formed on the support surface depending on the concentration used. This modified surface architecture orchestrates consequential alterations in the electronic and geometric configurations of the active metal, concurrently influencing catalytic performance. In this study, MoOx species were introduced into Pd in a controlled manner to substantially enhance the dehydrogenation activity of the N-heterocyclic liquid organic hydrogen carrier (LOHC) system. Pd-MoOx/Al2O3 catalyst—featuring an optimal 0.18 wt% Mo loading—demonstrated noteworthy improvement in activity, surpassing Pd/Al2O3 by a factor of 1.57 and 1.24, accompanied by enhanced recyclability in two distinct LOHC systems. Integrated characterization and theoretical calculations elucidated the modification of the electronic properties of both Pd and Al2O3 upon MoOx introduction and the corresponding adsorption behavior of the reactants, highlighting the charge transfer phenomenon from Pd to MoOx.
Mo loading—demonstrated noteworthy improvement in activity, surpassing Pd/Al2O3 by a factor of 1.57 and 1.24, accompanied by enhanced recyclability in two distinct LOHC systems. Integrated characterization and theoretical calculations elucidated the modification of the electronic properties of both Pd and Al2O3 upon MoOx introduction and the corresponding adsorption behavior of the reactants, highlighting the charge transfer phenomenon from Pd to MoOx.
111. Elucidation of Ce/Zr ratio effects on the physical properties and catalytic performance of CuOx/CeyZr1−yO2 catalysts
Journal paperAlthough cerium oxide (CeO2) is widely used as a catalyst support, its limited defect sites and surface oxygen vacancy/mobility should be improved. The incorporation of zirconium (Zr) in the cerium (Ce) lattice is shown to increase the number of oxygen vacancies and improve catalytic activity. Using a fixed surface density (SD) of copper (∼2.3 Cu atoms per nm2) as a surface species, the role of the support (CeyZr1−yO2 (y = 1.0, 0.9, 0.6, 0.5, and 0.0)) and defect site effects in the CO oxidation reaction was investigated. Spectroscopic (e.g., Raman, XRD, XPS) and microscopic (e.g., SEM-EDX, HR-TEM) characterization techniques were applied to evaluate the defect sites, crystallite size, lattice parameters, chemical composition, oxidation states of elements and microstructure of the catalysts. The CO oxidation reaction with varied CO : O2 ratios (1 : 5, 1 : 1, and 1 : 0.5 (stoichiometric)) was used as a model reaction to describe the relationship between the structure and the catalytic performance of each catalyst. Based on the characterization results of CeyZr1−yO2 materials, the addition of Zr causes physical and chemical changes to the overall material. The inclusion of Zr into the structure of CeO2 decreased the overall lattice parameter of the catalyst and increased the number of defect sites. The prepared catalysts were able to reach complete CO conversion  (∼100%) at low temperature conditions (<200 °C), each showing varied reaction activity. The difference in CO oxidation activity was then analyzed and related to the structure, wherein Cu loading, surface oxygen vacancies, reduction–oxidation ability, CuOx–support interaction and oxygen mobility in the catalyst were the crucial descriptors.
(∼100%) at low temperature conditions (<200 °C), each showing varied reaction activity. The difference in CO oxidation activity was then analyzed and related to the structure, wherein Cu loading, surface oxygen vacancies, reduction–oxidation ability, CuOx–support interaction and oxygen mobility in the catalyst were the crucial descriptors.
110. The Role of Size and Structure of Catalytic Active Sites in Polyolefin Hydrogenolysis
Journal paperThe increasing amount of plastic waste poses serious environmental problems that threaten both ecosys tems and human well-being. Hydrogenolysis has been widely studied as an effective approach for converting polyolefins into high-value liquids and waxy fuels. Their multifaceted reaction mechanism, including dehydrogenation, C–C bond cleavage, and hydrogenation, highlights the need for sophisticated catalyst design. The suppression of methane production, a persistent challenge in polyolefin hydrogenolysis, requires precise control of the cleavage site and inhibition of successive C–C bond cleavage. This delicate balance is achieved by carefully tuning the size and structure of metals. In this review, we investigate the effects of the size and structure of active sites on their catalytic activity and selectivity for the hydrogenolysis of polyolefins, including polyethylene (PE) and polypropylene (PP). A fundamental understanding of hydrogenolysis mechanisms, combined with strategic synthetic methodologies, is crucial for creating efficient catalysts with tailored properties.
tems and human well-being. Hydrogenolysis has been widely studied as an effective approach for converting polyolefins into high-value liquids and waxy fuels. Their multifaceted reaction mechanism, including dehydrogenation, C–C bond cleavage, and hydrogenation, highlights the need for sophisticated catalyst design. The suppression of methane production, a persistent challenge in polyolefin hydrogenolysis, requires precise control of the cleavage site and inhibition of successive C–C bond cleavage. This delicate balance is achieved by carefully tuning the size and structure of metals. In this review, we investigate the effects of the size and structure of active sites on their catalytic activity and selectivity for the hydrogenolysis of polyolefins, including polyethylene (PE) and polypropylene (PP). A fundamental understanding of hydrogenolysis mechanisms, combined with strategic synthetic methodologies, is crucial for creating efficient catalysts with tailored properties.
109. Enhanced Isoparaffin Selectivity in CO2 Hydrogenation by Combining Na-promoted Fe3O4 and Pt/WO3-ZrO2 Catalysts
Journal paperCatalytic CO2 hydrogenation facilitated by Fe3O4 produces long-chain hydrocarbons and light olefins through the combination of the reverse water-gas shift reaction and Fischer-Tropsch synthesis in a single reactor. Complementing Fe3O4-based catalysts, zeolites can be incorporated to modulate the product distribution of long-chain hydrocarbons in a dual-bed configuration. However, zeolites with strong acid sites induce the cracking of extended hydrocarbon chains, thereby impeding the production of hydrocarbons exceeding 12 carbon atoms. This paper introduces a novel Pt/WO3-ZrO2 (PtWZ) catalyst to produce liquid isoparaffins, aligning with the requirements of sustainable fuel characteristics. The precise adjustment of Pt and W contents in PtWZ enhances the hydrogenation and isomerization of linear olefins, amplifying the yield of isoparaffins. A dual-bed reactor integrating PtWZ (0.01 wt% Pt) with Na-promoted Fe3O4 catalyst achieved a CO2 conversion rate of 40% and a CO selectivity of 10%, while maximizing isomerization performance, attaining 42% isoparaffinic selectivity within liq uid hydrocarbons at operating conditions of 340 °C, 20 bar, WHSV = 4,500 mL·h−1·gcat−1. In contrast to zeolite catalysts prone to coke deposition during the reaction, the PtWZ catalyst exhibited remarkable stability, sustained activity without discernible deterioration, and negligible coke deposition, even after continuous operation for more than 100 h. This outcome delineates an optimized catalyst technology for the sustainable production of fuel from CO2, offering a viable alternative to the prevailing dependence on fossil fuels.
uid hydrocarbons at operating conditions of 340 °C, 20 bar, WHSV = 4,500 mL·h−1·gcat−1. In contrast to zeolite catalysts prone to coke deposition during the reaction, the PtWZ catalyst exhibited remarkable stability, sustained activity without discernible deterioration, and negligible coke deposition, even after continuous operation for more than 100 h. This outcome delineates an optimized catalyst technology for the sustainable production of fuel from CO2, offering a viable alternative to the prevailing dependence on fossil fuels.
108. Fundamental Structural Study of Hexagonal Boron Nitride (h-BN) and Boron Nitride Nanotube (BNNT) at Low and High Temperatures
Journal paperThe molecular structure stability at low and high temperature is important for an industrial application. The boron nitride-based materials, such as hexagonal boron nitride (h-BN) and boron nitride nanotubes (BNNTs), have been interested due to their high oxidation resistance and thermal stability. In this study, ex-situ and in-situ characterization techniques (e.g., Raman spectroscopy, X-ray Diffraction (XRD), and Fourier-transform infrared spectroscopy (FTIR)) were applied to investigate the structural change of BNNT and h-BN at high (up to 800°C) and low (down to -50 °C) temperatures. The Raman spectroscopy results showed that at high temperatures (800 °C), h-BN exhibited a significant red shift under both inert and oxidizing conditions, while BNNT showed no peak shift, indicating its more stable structural resistance compared to h-BN. Both h-BN and BNNT showed no peak shift after cooling to low temperatures (-50 °C). Stability of h-BN and BNNT up to a high temperature of 800 °C was revealed from the thermogravimetric analysis (TGA) and FTIR spectroscopy results. The FTIR results also indicate that under oxidizing conditions, heating h-BN results in the formation of more hydroxyl groups compared to BNNT. The in-situ XRD results showed a greater magnitude of lower 2θ shift with increasing temperatures for h-BN compared to BNNT. Additionally, there was a more significant increase in FWHM values with respect to temperatures for h-BN than BNNT regardless of the sample under inert or oxidizing conditions. The characterization results from this study indicate that BN-based materials, especially BNNT, are suitable candidates for high temperature chemical reaction applications.
The FTIR results also indicate that under oxidizing conditions, heating h-BN results in the formation of more hydroxyl groups compared to BNNT. The in-situ XRD results showed a greater magnitude of lower 2θ shift with increasing temperatures for h-BN compared to BNNT. Additionally, there was a more significant increase in FWHM values with respect to temperatures for h-BN than BNNT regardless of the sample under inert or oxidizing conditions. The characterization results from this study indicate that BN-based materials, especially BNNT, are suitable candidates for high temperature chemical reaction applications.
107. Atomically dispersed Rh catalysts formed on defective CeO2 surfaces with hydroformylation activity
Journal paperThe electron density of the metal active sites depends on the surface properties of the support. This study ai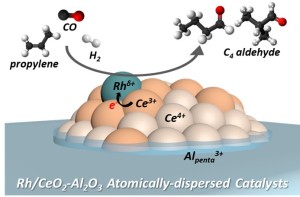 med to demonstrate that the formation of atomically dispersed Rh on ceria and the corresponding Rh electron density can be tuned depending on ceria concentration. The highly optimized Rh/CeO2-Al2O3 catalyst exhibited the highest activity during propylene hydroformylation, which was similar to that of the commercially available homogeneous catalyst RhCl(PPh3)3. Combined analytical results and theoretical calculations demonstrated that the increased Ce3+ fraction promoted high Rh electron density and attenuated CO adsorption strength, resulting in an increased hydroformylation activity. Additionally, the catalyst achieved separation and high added value by converting olefins to aldehydes through hydroformylation of olefins in a mixed C2‒C4 olefin/paraffin gas. This study showed that atomically dispersed metal supported catalysts can be designed as recyclable heterogeneous catalysts by tailoring the local environment and corresponding electron density of the metal to achieve high activity similar to that of the homogeneous catalysts that manipulate ligands around the metal.
med to demonstrate that the formation of atomically dispersed Rh on ceria and the corresponding Rh electron density can be tuned depending on ceria concentration. The highly optimized Rh/CeO2-Al2O3 catalyst exhibited the highest activity during propylene hydroformylation, which was similar to that of the commercially available homogeneous catalyst RhCl(PPh3)3. Combined analytical results and theoretical calculations demonstrated that the increased Ce3+ fraction promoted high Rh electron density and attenuated CO adsorption strength, resulting in an increased hydroformylation activity. Additionally, the catalyst achieved separation and high added value by converting olefins to aldehydes through hydroformylation of olefins in a mixed C2‒C4 olefin/paraffin gas. This study showed that atomically dispersed metal supported catalysts can be designed as recyclable heterogeneous catalysts by tailoring the local environment and corresponding electron density of the metal to achieve high activity similar to that of the homogeneous catalysts that manipulate ligands around the metal.
106. Tuning CuMgAl–Layered Double Hydroxide Nanostructures to Achieve CH4 and C2+ Product Selectivity in CO2 Electroreduction
Journal paperElectrochemical CO2 reduction reaction (eCO2RR) over Cu-based catalysts is a promising approach for efficiently converting CO2 into value-added chemicals and alternative fuels. However, achieving controllable product selectivity from eCO2RR remains challenging because of the difficulty in controlling the oxidation states of Cu against robust structural reconstructions during the eCO2RR. Herein, we report a novel strategy for tuning the oxidation states of Cu species a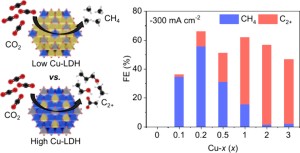 nd achieving eCO2RR product selectivity by adjusting the Cu content in CuMgAl-layered double hydroxide (LDH)-based catalysts. In this strategy, the highly stable Cu2+ species in low-Cu-containing LDHs facilitated the strong adsorption of *CO intermediates and further hydrogenation into CH4. Conversely, the mixed Cu0/Cu+ species in high-Cu-containing LDHs derived from the electroreduction during the eCO2RR accelerated C–C coupling reactions. This strategy to regulate Cu oxidation states using LDH nanostructures with low and high Cu molar ratios produced an excellent eCO2RR performance for CH4 and C2+ products, respectively.
nd achieving eCO2RR product selectivity by adjusting the Cu content in CuMgAl-layered double hydroxide (LDH)-based catalysts. In this strategy, the highly stable Cu2+ species in low-Cu-containing LDHs facilitated the strong adsorption of *CO intermediates and further hydrogenation into CH4. Conversely, the mixed Cu0/Cu+ species in high-Cu-containing LDHs derived from the electroreduction during the eCO2RR accelerated C–C coupling reactions. This strategy to regulate Cu oxidation states using LDH nanostructures with low and high Cu molar ratios produced an excellent eCO2RR performance for CH4 and C2+ products, respectively.
105. Laboratory-scale plastic upcycling and green growth: Evaluating the upcycling of plastic waste into carbon nanotubes from economic and environmental aspects
Journal paperIt is known that gas generated through plastic pyrolysis is abundant in hydrocarbons and can serve as a raw material for carbon nanotubes (CNTs). However, there has been no research evaluating both the economic and environmental feasibility of an upcycling process using pyrolysis gas to produce CNTs. A comparative evaluation was conducted between the upcycling process and the conventional process using methane as a carbon source, and each process was performed at l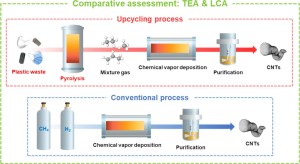 aboratory scale. We report that the cost of the upcycling process (2,999 $ kg−1) is comparable to that of the conventional process (2,930 $ kg−1), and that this slight cost disadvantage can be mitigated by reducing the Ar flow rate by 20%. Upcycling process involving pyrolysis has a greater impact on climate change than conventional process. However, this disadvantage can be significantly mitigated up to 19.38 kgCO2eq by substituting the power generation method with renewable energy source. Enhancing the prerequisites and technological maturity of upcycling processes can contribute to the expansion of the pyrolysis industry and the realization of a circular economy, thereby promoting green growth.
aboratory scale. We report that the cost of the upcycling process (2,999 $ kg−1) is comparable to that of the conventional process (2,930 $ kg−1), and that this slight cost disadvantage can be mitigated by reducing the Ar flow rate by 20%. Upcycling process involving pyrolysis has a greater impact on climate change than conventional process. However, this disadvantage can be significantly mitigated up to 19.38 kgCO2eq by substituting the power generation method with renewable energy source. Enhancing the prerequisites and technological maturity of upcycling processes can contribute to the expansion of the pyrolysis industry and the realization of a circular economy, thereby promoting green growth.
104. Ion-Exchange Desalination Battery with Reversible Chloride Capture
Journal paperSeawater desalination technology aims to address the global water shortage resulting from climate change. However, for a viabl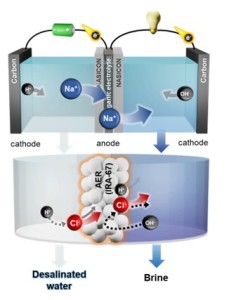 e solution, it is imperative to utilize renewable energy sources without carbon emissions. Desalination batteries represent a promising water–energy nexus technology; nonetheless, the limited reversibility of Cl– capture hinders their practicality. In this study, an ion-exchange desalination battery (IEDB), composed of an anion-exchange resin (AER) column filled with IRA-67 and a seawater battery (SWB), concurrently stores energy and desalinates seawater. During the charging of the SWB, Cl– are adsorbed on IRA-67, leading to seawater desalination. Subsequently, the Cl– ions are desorbed during discharge, automatically regenerating IRA-67 through the chemical potential. The proposed IEDB demonstrates 358 mL of seawater desalination with 1.48 Wh of energy storage through repetitive charge–discharge. This result secures stability of desalination batteries, which is essential for efficient water-energy nexus.
e solution, it is imperative to utilize renewable energy sources without carbon emissions. Desalination batteries represent a promising water–energy nexus technology; nonetheless, the limited reversibility of Cl– capture hinders their practicality. In this study, an ion-exchange desalination battery (IEDB), composed of an anion-exchange resin (AER) column filled with IRA-67 and a seawater battery (SWB), concurrently stores energy and desalinates seawater. During the charging of the SWB, Cl– are adsorbed on IRA-67, leading to seawater desalination. Subsequently, the Cl– ions are desorbed during discharge, automatically regenerating IRA-67 through the chemical potential. The proposed IEDB demonstrates 358 mL of seawater desalination with 1.48 Wh of energy storage through repetitive charge–discharge. This result secures stability of desalination batteries, which is essential for efficient water-energy nexus.

103. Insight into the Synergistic Effect of the Oxide−Metal Interface on Hot Electron Excitation
Journal paper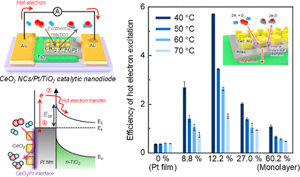 Formulating a quantitative relationship between the extent of electron transferat metal−oxide interfaces and catalytic performance aids the rational designof oxide-supported metal catalysts. An effective strategy for monitoring electron transfer at nanoscale interfacial sites is to detect in real time the hot electrons excited when catalytic reactions occur at metal−oxide perimeter sites.Here, based on our in situ techniques for extracting electron transfer as a currentsignal using a catalytic nanodiode sensor, we observe hot electron excitation at the CeO2/Pt interface during H2 oxidation. By quantitatively analyzing the hot electrons released during the reaction,we identified the optimal concentration of CeO2/Pt interfaces that maximize the catalytic performance of CeO2/Pt. Through a combinatorial study of experiment and theory,we confirm the decisive role of CeO2/Pt interfacial sites in improving the reactivity and electronic excitation.
Formulating a quantitative relationship between the extent of electron transferat metal−oxide interfaces and catalytic performance aids the rational designof oxide-supported metal catalysts. An effective strategy for monitoring electron transfer at nanoscale interfacial sites is to detect in real time the hot electrons excited when catalytic reactions occur at metal−oxide perimeter sites.Here, based on our in situ techniques for extracting electron transfer as a currentsignal using a catalytic nanodiode sensor, we observe hot electron excitation at the CeO2/Pt interface during H2 oxidation. By quantitatively analyzing the hot electrons released during the reaction,we identified the optimal concentration of CeO2/Pt interfaces that maximize the catalytic performance of CeO2/Pt. Through a combinatorial study of experiment and theory,we confirm the decisive role of CeO2/Pt interfacial sites in improving the reactivity and electronic excitation.
102. Nanoscale Precursor Distribution by Microfluidization for Scalable Production of Highly Efficient Thermocatalysts
Journal paperThe preparation of two-dimensional (2D) materials often requires complicated exfoliation procedures having low yields. The exfolia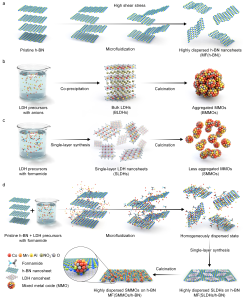 ted nanosheets are prone to thermal aggregation and unsuitable for thermocatalysis. Herein, a scalable approach produced 2D catalyst precursors well-distributed and mixed at the nanoscale. Using continuous microfluidization and single-layer layered double hydroxide (LDH) synthesis, the prepared suspension contained exfoliated hexagonal boron nitride (h-BN) nanosheets and single-layer LDHs. The increased contact area between h-BN and LDHs enabled the formation of highly dispersed MnCoAl mixed metal oxide nanoparticles anchored on h-BN nanosheets after calcination. In the selective catalytic reduction of NOx with NH3 (NH3-SCR, a representative thermocatalytic application), this nanocomposite demonstrated a record turnover frequency of 0.772 h-1 among reported Mn-based NH3-SCR catalysts, with high NOx conversion and high N2 selectivity at low temperatures. By creating 2D precursors mixed at the nanoscale, this new synthetic approach can realize the scalable production of highly efficient thermocatalysts.
ted nanosheets are prone to thermal aggregation and unsuitable for thermocatalysis. Herein, a scalable approach produced 2D catalyst precursors well-distributed and mixed at the nanoscale. Using continuous microfluidization and single-layer layered double hydroxide (LDH) synthesis, the prepared suspension contained exfoliated hexagonal boron nitride (h-BN) nanosheets and single-layer LDHs. The increased contact area between h-BN and LDHs enabled the formation of highly dispersed MnCoAl mixed metal oxide nanoparticles anchored on h-BN nanosheets after calcination. In the selective catalytic reduction of NOx with NH3 (NH3-SCR, a representative thermocatalytic application), this nanocomposite demonstrated a record turnover frequency of 0.772 h-1 among reported Mn-based NH3-SCR catalysts, with high NOx conversion and high N2 selectivity at low temperatures. By creating 2D precursors mixed at the nanoscale, this new synthetic approach can realize the scalable production of highly efficient thermocatalysts.
101. Metallic nickel exsolved from a two-dimensional MWW-type zeolitic nickel silicate: An effective catalyst for ammonia decomposition
Journal paperExsolution of active species from metal oxide materials is an effective strategy for preparing a highly active and stable catalyst. Here, we prepared two-dimensional nickel (Ni) silicate material with delaminated MWW layers (Ni-DMLs) by hydrotherma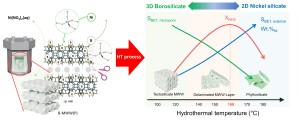 l treatment of borosilicate MWW precursor (B-MWW(P)) with Ni nitrate solutions and applied on the catalytic decomposition of ammonia (NH3) via exsolution. The layered B-MWW(P) was transformed to a three-dimensional (3D) tectosilicate MWW, a two-dimensional (2D) Ni-DML, and a 2D phyllosilicate structures at temperature regions of 100–120 °C, 140–160 °C, and 170–180 °C, respectively. Meanwhile, the Ni contents on the samples increased from 3.6 to 37.6 wt.% as the hydrothermal temperature increased from 100 to 180 °C, owing to the substitution of framework B by Ni. The chemical state of Ni on each sample was characterized by various analytical tools and correlated with their catalytic properties of NH3 decomposition over the exsolved metallic Ni species. The NH3 decomposition over Ni-DMLs was evaluated in the temperature range of 300–600 °C, and the apparent activation energies were compared. The Ni-DML-160 exhibited the best catalytic activity, achieving an NH3 conversion of 70% at 500 °C and maintained 90% to initial conversion during 100 h on stream at 550 °C. The synergistic effect of the strong interaction between the exsolved metallic Ni and the zeolite support and the 2D nature of Ni-DMLs relieved the catalytic deactivation by sintering and coking, respectively.
l treatment of borosilicate MWW precursor (B-MWW(P)) with Ni nitrate solutions and applied on the catalytic decomposition of ammonia (NH3) via exsolution. The layered B-MWW(P) was transformed to a three-dimensional (3D) tectosilicate MWW, a two-dimensional (2D) Ni-DML, and a 2D phyllosilicate structures at temperature regions of 100–120 °C, 140–160 °C, and 170–180 °C, respectively. Meanwhile, the Ni contents on the samples increased from 3.6 to 37.6 wt.% as the hydrothermal temperature increased from 100 to 180 °C, owing to the substitution of framework B by Ni. The chemical state of Ni on each sample was characterized by various analytical tools and correlated with their catalytic properties of NH3 decomposition over the exsolved metallic Ni species. The NH3 decomposition over Ni-DMLs was evaluated in the temperature range of 300–600 °C, and the apparent activation energies were compared. The Ni-DML-160 exhibited the best catalytic activity, achieving an NH3 conversion of 70% at 500 °C and maintained 90% to initial conversion during 100 h on stream at 550 °C. The synergistic effect of the strong interaction between the exsolved metallic Ni and the zeolite support and the 2D nature of Ni-DMLs relieved the catalytic deactivation by sintering and coking, respectively.
100. Soccer Ball-like Assembly of Edge-to-edge Oriented 2D-silica Nanosheets: A Promising Catalyst Support for High-Temperature Reforming
Journal paperControlled assembly of nanoparticles into well-defined assembled architectures th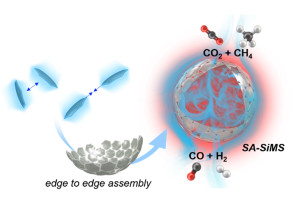 rough precise manipulation of spatial arrangement and interactions allows the development of advanced mesoscale materials with tailored structures, hierarchical functionalities, and enhanced properties. Despite remarkable advancements, the controlled assembly of highly anisotropic 2Dnanosheets is significantly challenging, primarily due to the limited availability of selective edge-to-edge connectivity compared to the abundant large faces.
rough precise manipulation of spatial arrangement and interactions allows the development of advanced mesoscale materials with tailored structures, hierarchical functionalities, and enhanced properties. Despite remarkable advancements, the controlled assembly of highly anisotropic 2Dnanosheets is significantly challenging, primarily due to the limited availability of selective edge-to-edge connectivity compared to the abundant large faces. Innovative strategies are needed to unlock the full potential of 2D-nanomaterialsin self-assembled structures with distinct and desirable properties. This research unveils the discovery of controlled self-assembly of 2D-silica nanosheets (2D-SiNSs) into hollow micron-sized soccer ball-like shells (SA-SiMS). The assembly is driven by the physical flexibility of the 2D-SiNSs and the differential electricdouble-layer charge gradient creating electrostatic bias on the edge and face regions. The resulting SA-SiMS structures exhibit high mechanical stability, even at high-temperatures, and exhibit excellent performance as catalyst support in the dry reforming of methane. The SA-SiMS structures facilitate improved mass transport, leading to enhanced reaction rates, while the thin silica shell prevents sintering of small catalyst nanocrystals, thereby preventing coke formation. This discovery sheds light on the controllable self-assembly of 2D nanomaterials and provides insights into the design and synthesis of advanced mesoscale materials with tailored properties.
Innovative strategies are needed to unlock the full potential of 2D-nanomaterialsin self-assembled structures with distinct and desirable properties. This research unveils the discovery of controlled self-assembly of 2D-silica nanosheets (2D-SiNSs) into hollow micron-sized soccer ball-like shells (SA-SiMS). The assembly is driven by the physical flexibility of the 2D-SiNSs and the differential electricdouble-layer charge gradient creating electrostatic bias on the edge and face regions. The resulting SA-SiMS structures exhibit high mechanical stability, even at high-temperatures, and exhibit excellent performance as catalyst support in the dry reforming of methane. The SA-SiMS structures facilitate improved mass transport, leading to enhanced reaction rates, while the thin silica shell prevents sintering of small catalyst nanocrystals, thereby preventing coke formation. This discovery sheds light on the controllable self-assembly of 2D nanomaterials and provides insights into the design and synthesis of advanced mesoscale materials with tailored properties.
99. Ceria Tubular Nanoarchitecture Antioxidants Achieve Sustainable Fuel Cell Devices via Tuning the Oxophilicity of Pt Catalytic Surfaces and Radical Scavenging
Journal paperThe durability of polymer electrolyte membrane fuel cells (PEMFCs) crucially depends on the use of antioxidants to prevent el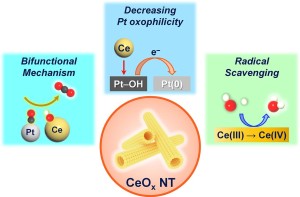 ectrocatalyst degradation. Here, we report for the first time, by in situ X-ray absorption and H2O2 electrochemistry, that ceria significantly weakens the Pt-surface oxophilicity, which determines oxygen reduction (ORR) activity and durability, within the PEMFC cathode. Ceria mitigates catalyst disintegration and improves ORR durability by Pt oxophilicity reduction and its inherent radical scavenging behavior. We also found that the antioxidation efficacy of ceria could be finely tuned through nanostructuring. Among various ceria nanostructures, tubular ceria nanoarchitectures (CeOx NT), designed to have the largest surface area and abundant oxygen vacancies, enable the most potent interaction between Pt and Ce without direct chemical contact with Pt. The nanotubular structure confers superior multifunctional antioxidant therapeutic efficacy to Pt/C catalyst in PEMFCs, resulting in outstanding durability that retains 94% of initial performance after 100-hour tests.
ectrocatalyst degradation. Here, we report for the first time, by in situ X-ray absorption and H2O2 electrochemistry, that ceria significantly weakens the Pt-surface oxophilicity, which determines oxygen reduction (ORR) activity and durability, within the PEMFC cathode. Ceria mitigates catalyst disintegration and improves ORR durability by Pt oxophilicity reduction and its inherent radical scavenging behavior. We also found that the antioxidation efficacy of ceria could be finely tuned through nanostructuring. Among various ceria nanostructures, tubular ceria nanoarchitectures (CeOx NT), designed to have the largest surface area and abundant oxygen vacancies, enable the most potent interaction between Pt and Ce without direct chemical contact with Pt. The nanotubular structure confers superior multifunctional antioxidant therapeutic efficacy to Pt/C catalyst in PEMFCs, resulting in outstanding durability that retains 94% of initial performance after 100-hour tests.
98. Reversible Pd Catalysts Supported on Hierarchical Titanate Nanosheets for an N-methylindole-based Liquid Organic Hydrogen Carrier
Journal paperReversible hydrogenation and dehydrogenation processes were investigated in a liquid organic hydrogen carrier (LOHC) system, employing a single-catalyst approach. Key hydrogen-involved catalytic behaviors, including adsorption and migration, play crucial roles in reactivity. To facilitate these behaviors at the active sites on the catalyst surface during the LOHC process, a defective metal oxide support was 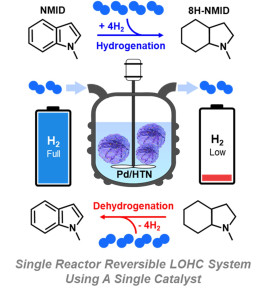 utilized. Herein, a Pd catalyst was prepared using hierarchical titanate nanosheets (HTN) synthesized via solvothermal synthesis. Compared to commercial TiO2 and hierarchical TiO2 (HT) which was synthesized by the calcination of HTN, HTN exhibited a higher density of acidic sites and oxygen vacancies. Density functional theory calculations confirmed that hydrogen spillover occurred more readily on the defective HTN surface than on the TiO2 (101) surface. The Pd/HTN catalyst demonstrated superior catalytic activity for both the hydrogenation and dehydrogenation reactions in the N-methylindole-based LOHC system. The hydrogen uptake of Pd/HTN catalyst (4.73 wt%) was three times higher than those of other Pd catalysts (~1.57 wt%). The single Pd/HTN catalyst successfully accomplished reversible hydrogen storage and release within the LOHC system in one reactor.
utilized. Herein, a Pd catalyst was prepared using hierarchical titanate nanosheets (HTN) synthesized via solvothermal synthesis. Compared to commercial TiO2 and hierarchical TiO2 (HT) which was synthesized by the calcination of HTN, HTN exhibited a higher density of acidic sites and oxygen vacancies. Density functional theory calculations confirmed that hydrogen spillover occurred more readily on the defective HTN surface than on the TiO2 (101) surface. The Pd/HTN catalyst demonstrated superior catalytic activity for both the hydrogenation and dehydrogenation reactions in the N-methylindole-based LOHC system. The hydrogen uptake of Pd/HTN catalyst (4.73 wt%) was three times higher than those of other Pd catalysts (~1.57 wt%). The single Pd/HTN catalyst successfully accomplished reversible hydrogen storage and release within the LOHC system in one reactor.
97. Efficient Fe3O4 Nanoparticle Catalysts for Depolymerization of Polyethylene Terephthalate
Journal paperPolyethylene terephthalate (PET) can be recovered as high-purity bis(2-hydroxyethyl terephthalate) (BHET) monomer by glycolysis in the presence of Fe3O4 nanoparticles (NPs). In this study, Fe3O4 NPs of various shapes, sizes, and surface areas were synthesized using different colloidal synthesis methods, and the conversion of PET glycolysis and BHET yield were compared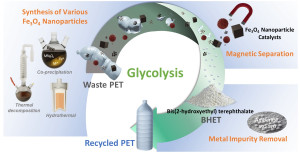 . Spinel ferrite NPs, including Fe3O4, were synthesized using the coprecipitation (CP), thermal decomposition (TD), and the hydrothermal (H) methods. Among the NP catalysts, Fe3O4-CP exhibited the best glycolysis performance with a PET conversion of ~100% and BHET yield of 93.5% at 195 °C for 2 h owing to its high surface area (146.6 m2g-1). The larger the surface area and the better the dispersion, the higher the glycolysis activity. The glycolysis performance of the mixed spinel ferrite NPs was similar to that of the Fe3O4 NPs, indicating that replacing Fe2+ in the Fe3O4 NPs with other transition metals, M2+, did not significantly change the glycolysis performance. BHET monomers produced from commercial waste PET bottles in large quantities contained trace amounts of metal contaminants, because PET production uses various metal-based additives and catalysts. Amberlite IRC-120, a cation-exchange resin, effectively removed metal impurities from BHET. This study provides an effective strategy for producing recycled PET (r-PET) by waste PET glycolysis.
. Spinel ferrite NPs, including Fe3O4, were synthesized using the coprecipitation (CP), thermal decomposition (TD), and the hydrothermal (H) methods. Among the NP catalysts, Fe3O4-CP exhibited the best glycolysis performance with a PET conversion of ~100% and BHET yield of 93.5% at 195 °C for 2 h owing to its high surface area (146.6 m2g-1). The larger the surface area and the better the dispersion, the higher the glycolysis activity. The glycolysis performance of the mixed spinel ferrite NPs was similar to that of the Fe3O4 NPs, indicating that replacing Fe2+ in the Fe3O4 NPs with other transition metals, M2+, did not significantly change the glycolysis performance. BHET monomers produced from commercial waste PET bottles in large quantities contained trace amounts of metal contaminants, because PET production uses various metal-based additives and catalysts. Amberlite IRC-120, a cation-exchange resin, effectively removed metal impurities from BHET. This study provides an effective strategy for producing recycled PET (r-PET) by waste PET glycolysis.
96. Upcycling of plastic waste into carbon nanotubes as efficient battery additives
Journal paperCarbon nanotubes (CNTs) were produced from waste face mask and non-recyclable mixed pl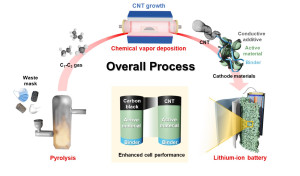 astic waste via pyrolysis-chemical vapor deposition (CVD). The yield and properties of the prepared CNTs depended on the feedstock and catalyst used. CoMo/MgO and FeMo/MgO were found to be suitable catalysts for producing few-walled and multi-walled CNTs, respectively, regardless of the feedstock. Both excellent carbon yield (516.7 wt.%) and CNT purity (97.9 wt.%) occurred when using the mask waste and FeMo/MgO catalyst. The resulting CNTs were mixed with LiNi0.8Co0.1Mn0.1O2
astic waste via pyrolysis-chemical vapor deposition (CVD). The yield and properties of the prepared CNTs depended on the feedstock and catalyst used. CoMo/MgO and FeMo/MgO were found to be suitable catalysts for producing few-walled and multi-walled CNTs, respectively, regardless of the feedstock. Both excellent carbon yield (516.7 wt.%) and CNT purity (97.9 wt.%) occurred when using the mask waste and FeMo/MgO catalyst. The resulting CNTs were mixed with LiNi0.8Co0.1Mn0.1O2  (NCM811) active material and poly(vinylidene fluoride) binder to fabricate cathodes. Electrochemical measurements showed that CNTs grown on the FeMo/MgO catalyst outperformed commercial carbon black and CNTs. Since C1‒C3 hydrocarbons and H2 in the plastic pyrolysis gas can be directly used for CNT production without gas separation or purification, the proposed pyrolysis-CVD process is favorable for efficient plastic upcycling and advanced battery applications.
(NCM811) active material and poly(vinylidene fluoride) binder to fabricate cathodes. Electrochemical measurements showed that CNTs grown on the FeMo/MgO catalyst outperformed commercial carbon black and CNTs. Since C1‒C3 hydrocarbons and H2 in the plastic pyrolysis gas can be directly used for CNT production without gas separation or purification, the proposed pyrolysis-CVD process is favorable for efficient plastic upcycling and advanced battery applications.
95. A-Site Effects of Titanate-Perovskite (ATiO3)-Based Catalysts on Dehydrogenation of N-Heterocyclic Molecules
Journal paperDehydrogenation reactions in liquid organic hydrogen carrier (LOHC) systems present significant challenges, particularly when aiming for low-temperature operations while ensuring that no hydrogen remains in the substrate molecules. Enhancing catalytic performance requires modifying the adsorption behavior of the reactants and products during dehydrogenation. Perovskites have emerged as promising catalyst supports because of their ability to modify the surface chemical properties by manipulating the cations present at the A and B sites. This study investigated the effects of A-site cations (Ca, Sr, and Ba) in titanate-type perovskite (ATiO3)—a prototypical perovskite—on the dehydrogenation activ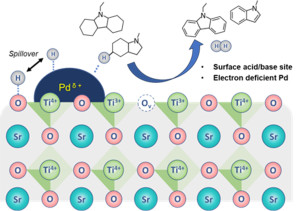 ity in an LOHC system. Remarkably, Pd/SrTiO3 exhibited outstanding performance by completely converting octahydro-N-methylindole to N-methylindole and releasing 5.76 wt% hydrogen over 8 h. Additionally, it dehydrogenated dodecahydro-N-ethylcarbazole to N-ethylcarbazole with a hydrogen release of 5.70 wt%. Furthermore, the catalyst demonstrated a stable performance after recycling tests for three times without degradation or loss of activity. The chemical state of the catalyst surface was characterized through X-ray photoelectron spectroscopy, H2-temperature programmed reduction, and chemisorption using NH3, CO2, and H2. The results revealed that the exceptional dehydrogenation activity of Pd/SrTiO3 is due to the presence of suitable surface oxygen vacancies and abundant acid–base sites.
ity in an LOHC system. Remarkably, Pd/SrTiO3 exhibited outstanding performance by completely converting octahydro-N-methylindole to N-methylindole and releasing 5.76 wt% hydrogen over 8 h. Additionally, it dehydrogenated dodecahydro-N-ethylcarbazole to N-ethylcarbazole with a hydrogen release of 5.70 wt%. Furthermore, the catalyst demonstrated a stable performance after recycling tests for three times without degradation or loss of activity. The chemical state of the catalyst surface was characterized through X-ray photoelectron spectroscopy, H2-temperature programmed reduction, and chemisorption using NH3, CO2, and H2. The results revealed that the exceptional dehydrogenation activity of Pd/SrTiO3 is due to the presence of suitable surface oxygen vacancies and abundant acid–base sites.
94. Coke resistant NiCo/CeO2 catalysts for dry reforming of methane derived from core@shell Ni@Co nanoparticles
Journal paperCore@shell Ni@Co and bimetallic alloyed Ni–Co nanoparticles with controlled Co/Ni compositions were prepared and supported on CeO2 to investigate their performance in catalytic dry reforming of methane (DRM) and occurrence of sintering and coking. Increasing the Co/Ni ratio significantly reduced coke deposition while maintaining catalytic activity for DRM. However, a Co/Ni ratio >1 caused a rapid decrease in activity. The Ni@Co1/CeO2 catalyst exhibited the highest CH4 and CO2 conversions, with long-term stability during DRM at 800 °C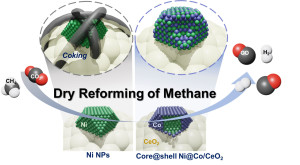 for 100 h. The initial core@shell structure of the Ni@Co1/CeO2 catalyst transformed to a homogeneous alloy after DRM at 800 °C for 10 h, losing its Co shell. However, the bimetallic alloyed Ni–Co1/CeO2 catalyst transformed into a non-uniform alloy rich in Co on the surface after DRM for 10 h. As the elemental distribution of the NPs becomes more homogeneous, Ni–Co1/CeO2 exhibit similar catalytic activity to Ni@Co1/CeO2 after 50 h. The oxygen vacancies on the CeO2 surface provided oxygen atoms to the Ni surface, removing carbon species deposited and releasing CO. Therefore, Ni@Co1/CeO2 catalyst provides excellent catalytic activity and stability due to the rapid formation of a homogenous alloy and the synergistic effect of Co and CeO2.
for 100 h. The initial core@shell structure of the Ni@Co1/CeO2 catalyst transformed to a homogeneous alloy after DRM at 800 °C for 10 h, losing its Co shell. However, the bimetallic alloyed Ni–Co1/CeO2 catalyst transformed into a non-uniform alloy rich in Co on the surface after DRM for 10 h. As the elemental distribution of the NPs becomes more homogeneous, Ni–Co1/CeO2 exhibit similar catalytic activity to Ni@Co1/CeO2 after 50 h. The oxygen vacancies on the CeO2 surface provided oxygen atoms to the Ni surface, removing carbon species deposited and releasing CO. Therefore, Ni@Co1/CeO2 catalyst provides excellent catalytic activity and stability due to the rapid formation of a homogenous alloy and the synergistic effect of Co and CeO2.
93. Enhancing Catalytic Performance and Hot Electron Generation through Engineering Metal-Oxide and Oxide-Oxide Interfaces
Journal paperThe role of interfaces in catalytic reactions is of utmost importance, influencing reaction kinetics and electron transfer processes. However, investigations in combined interfaces of metal-oxide and oxide-oxide at heterogeneous catalysts still have challenges due to their complex structure. Herein, we synthesized well-defined Co3O4 and CeO2 cubes with distinct facets and investigated their catalytic performance when deposited on a Pt-thin film, focusing on the influence of metal-oxide and oxide-oxide interfaces. Catalytic measurements demonstrated that the CeO2/Pt interface significantly enhanced turnover frequency (TOF) and selectivity for partial methanol oxidation compared to Co3O4/Pt and bare Pt. Notably, the CeO2/Co3O4/Pt nanodevice exhibited improved partial oxidation selectivity, highlighting the role of the CeO2/Co3O4 interface in methy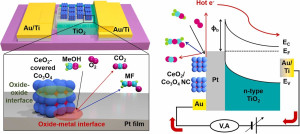 l formate production. Chemicurrent measurements demonstrate enhanced hot electron generation due to increased overall TOF and partial oxidation production. We also conducted near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) analysis, revealing a higher concentration of Ce3+ ions and increased oxygen vacancies in the CeO2/Co3O4/Pt catalyst, suggesting oxygen migration from CeO2 to Co3O4, leading to methoxy species stabilization and promoting methyl formate formation.
l formate production. Chemicurrent measurements demonstrate enhanced hot electron generation due to increased overall TOF and partial oxidation production. We also conducted near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) analysis, revealing a higher concentration of Ce3+ ions and increased oxygen vacancies in the CeO2/Co3O4/Pt catalyst, suggesting oxygen migration from CeO2 to Co3O4, leading to methoxy species stabilization and promoting methyl formate formation.
92. Hydrogen production by the catalytic decomposition of ammonia over a Ru/SiCeOx catalyst: The synergistic effect of Si addition
Journal paperControlling active metal–support interaction is critical in the catalytic decomposition of ammonia (NH3). In this study, we investigated the generation of oxygen vacancies in the SiCeOx support by Si addition and the increase in catalytic performance for the catalytic decomposition of NH3 by their interaction with Ru. The Si content in the Ru/SiCeOx catalysts was controlled by glassware elution during the 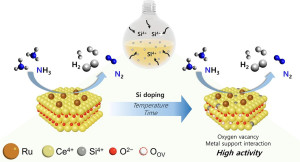 precipitation of Ce(NO3)3 at a high pH level (10.5) by applying different aging temperatures and times. The generation of oxygen vacancies by the insertion of Si4+ into the CeO2 lattice was characterized by O2-pulse experiments and Raman and X-ray photoelectron spectroscopy analyses. The influence of oxygen vacancies on the adsorption strengths of hydrogen and nitrogen on Ru was characterized by H2-temperature-programmed reduction and NH3-temperature-programmed desorption. The correlation between NH3 conversion, oxygen vacancy content, and nitrogen desorption temperature was experimentally proven.
precipitation of Ce(NO3)3 at a high pH level (10.5) by applying different aging temperatures and times. The generation of oxygen vacancies by the insertion of Si4+ into the CeO2 lattice was characterized by O2-pulse experiments and Raman and X-ray photoelectron spectroscopy analyses. The influence of oxygen vacancies on the adsorption strengths of hydrogen and nitrogen on Ru was characterized by H2-temperature-programmed reduction and NH3-temperature-programmed desorption. The correlation between NH3 conversion, oxygen vacancy content, and nitrogen desorption temperature was experimentally proven.
91. Crystallinity-modulated hollow CeO2-x nanorods as free radical scavengers for long-term photostability in organic photovoltaics
Journal paperHere we investigated the effects of CeO2−x nanostructures as free radical scavengers on the long-term photostability of an organic photovoltaic (OPV) structure. From powder X-ray diffraction, Raman spectroscopy, X-ray photoelectron spectroscopy, and N2 adsorption experime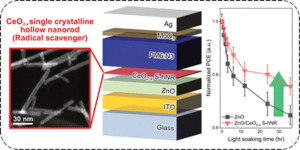 nts, it was determined that the single-crystalline hollow CeO2−x nanorods were very effective as hydroxyl radical scavengers. This was attributed to their having more Ce3+ states and a wider surface area than other types of CeO2 nanostructures. Time-dependent UV-visible absorption spectra analyses also revealed that the improved scavenging of hydroxyl radicals in the OPV device was related to the better interfacial compatibility between the organic active and ZnO layers, resulting in improved OPV photostability.
nts, it was determined that the single-crystalline hollow CeO2−x nanorods were very effective as hydroxyl radical scavengers. This was attributed to their having more Ce3+ states and a wider surface area than other types of CeO2 nanostructures. Time-dependent UV-visible absorption spectra analyses also revealed that the improved scavenging of hydroxyl radicals in the OPV device was related to the better interfacial compatibility between the organic active and ZnO layers, resulting in improved OPV photostability.
90. Zeolitic Imidazolate Framework Decorated Molybdenum Carbide Catalysts for Hydrodeoxygenation of Guaiacol to Phenol
Journal paper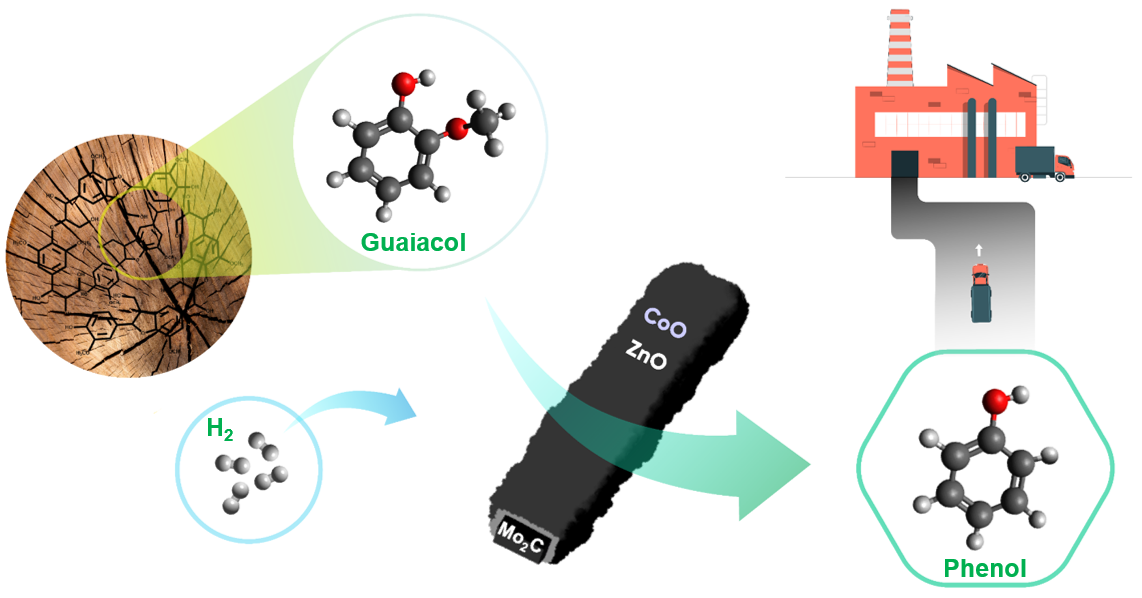 Bimetallic zeolitic imidazolate framework (BMZIF)-decorated Mo carbide catalysts were designed for the catalytic hydrodeoxygenation of guaiacol to produce phenol with high selectivity. A uniform layer of BMZIF was systematically coated onto the surface of the MoO3 nanorods. During carbonization at 700 °C for 4 h, BMZIF generated active species (ZnO, CoO) on highly dispersed N-doped carbons, creating a porous shell structure. Simultaneously, the MoO3 nanorod was transformed into the Mo2C phase. The resulting core@shell type Mo2C@BMZIF-700 °C (4 h) catalyst promoted a 97% guaiacol conversion and 70% phenol selectivity under 4 MPa of H2 at 330 °C for 4 h, which was not achieved by other supported catalysts. The catalyst also showed excellent selective cleavage of the methoxy group of lignin derivatives (syringol and vanillin), which makes it suitable for selective demethoxylation in future biomass catalysis. Moreover, it exhibits excellent recyclability and stability without changing the structure or active species.
Bimetallic zeolitic imidazolate framework (BMZIF)-decorated Mo carbide catalysts were designed for the catalytic hydrodeoxygenation of guaiacol to produce phenol with high selectivity. A uniform layer of BMZIF was systematically coated onto the surface of the MoO3 nanorods. During carbonization at 700 °C for 4 h, BMZIF generated active species (ZnO, CoO) on highly dispersed N-doped carbons, creating a porous shell structure. Simultaneously, the MoO3 nanorod was transformed into the Mo2C phase. The resulting core@shell type Mo2C@BMZIF-700 °C (4 h) catalyst promoted a 97% guaiacol conversion and 70% phenol selectivity under 4 MPa of H2 at 330 °C for 4 h, which was not achieved by other supported catalysts. The catalyst also showed excellent selective cleavage of the methoxy group of lignin derivatives (syringol and vanillin), which makes it suitable for selective demethoxylation in future biomass catalysis. Moreover, it exhibits excellent recyclability and stability without changing the structure or active species.
89. Versatile Layered Hydroxide Precursors for Generic Synthesis of Cu-Based Materials
Journal paperThe ability to mix multiple elements in a structure is crucial for obtaining Cu-based nanostructures and microstructu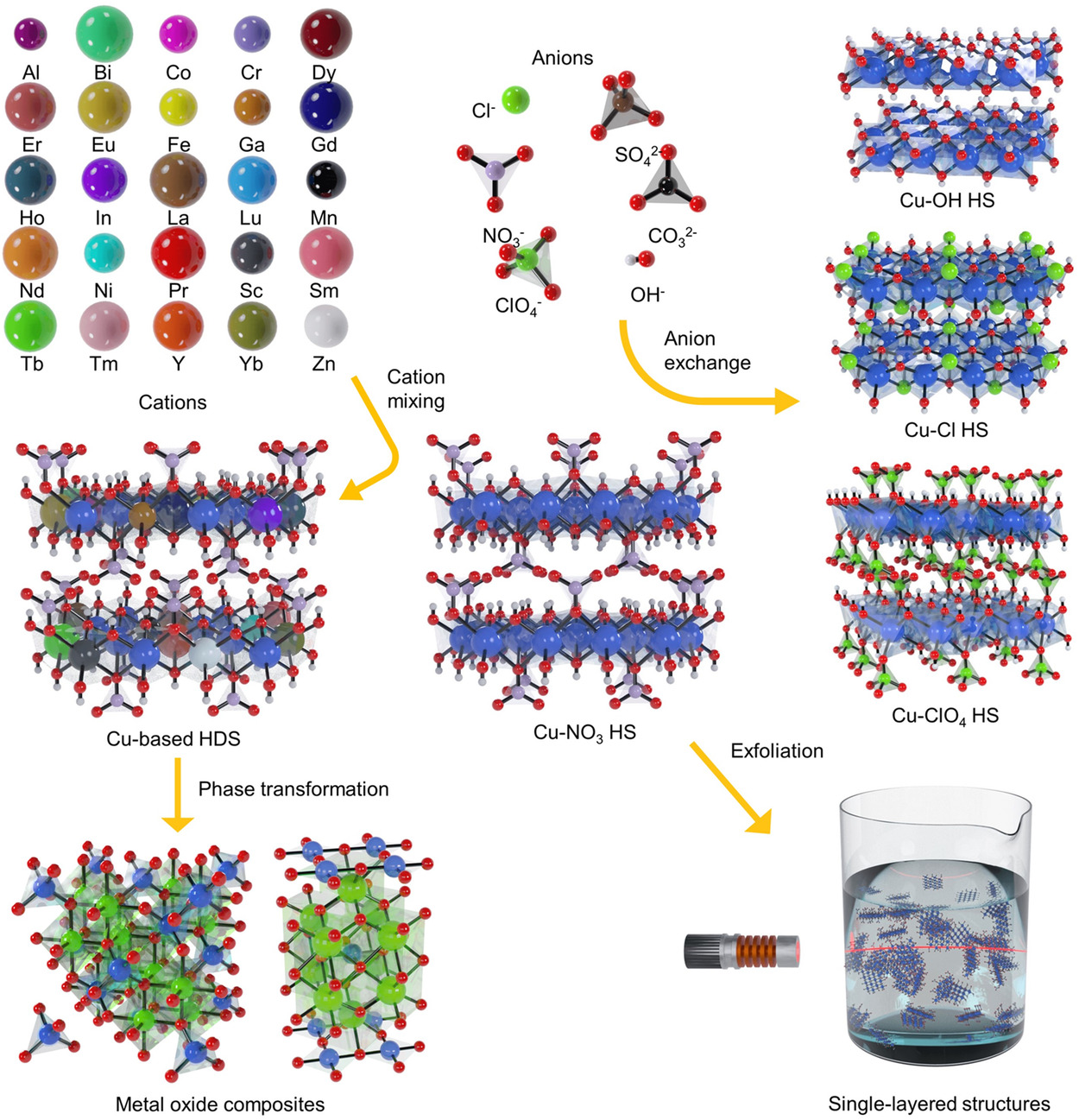 res with desirable physicochemical properties. Precursors containing multiple metal cations, such as layered double hydroxides, have been used for the synthesis of multielement materials. However, these precursors experience difficulty containing large cations, which limits the functionalities of the derived materials. Herein, the development of Cu-based hydroxy double salts (HDSs), versatile precursors that accommodate a broad range of metal cations with homogeneous distributions, is reported. Up to 25 different metal cations are mixed with Cu in an HDS single phase, individually and simultaneously. The HDSs further exhibit useful properties with respect to anion exchange, exfoliation, and phase transformation to metal oxides. During the metal oxide transformation process, the formed crystal structures are mainly dependent on the ionic radius of the secondary metal cations. To prove their utility as precursors, the metal oxides derived from the HDSs are tested and found that they exhibited high catalytic activities for CO oxidation. This study significantly expands the compositional and structural freedom of Cu-based multi-elemental materials.
res with desirable physicochemical properties. Precursors containing multiple metal cations, such as layered double hydroxides, have been used for the synthesis of multielement materials. However, these precursors experience difficulty containing large cations, which limits the functionalities of the derived materials. Herein, the development of Cu-based hydroxy double salts (HDSs), versatile precursors that accommodate a broad range of metal cations with homogeneous distributions, is reported. Up to 25 different metal cations are mixed with Cu in an HDS single phase, individually and simultaneously. The HDSs further exhibit useful properties with respect to anion exchange, exfoliation, and phase transformation to metal oxides. During the metal oxide transformation process, the formed crystal structures are mainly dependent on the ionic radius of the secondary metal cations. To prove their utility as precursors, the metal oxides derived from the HDSs are tested and found that they exhibited high catalytic activities for CO oxidation. This study significantly expands the compositional and structural freedom of Cu-based multi-elemental materials.
88. Complete utilization of waste lignin: preparation of lignin-derived carbon supports and conversion of lignin-derived guaiacol to nylon precursors
Journal paperThe valorization of waste lignin for the production of high value-added chemicals is energetically and environmentally important. In this study, a new catalytic process was developed to produce raw materials for nylon production utilizing 100% of waste lignin emitted from industrial processes. Guaiacol, extracted from technical lignin, was converted to phenol through a hydrodeoxygenation reaction over carbon-supported MoO2 catalysts. The extracted lignin oil served as a carbon source to prepare lignin-derived porous carbons possessing an interconnected porous structure with a large pore size and volume via a nanocasting method. The high dispersion of MoO2 deposited on the porous carbon support enables a high guaiacol conversion (98.4%) and high phenol selectivity (73.7%) compared to those of a typical activated carbon support. The resulting phenol was selectively converted to cyclohexanone or cyclohexanol, depending on the controlled hydrogenation environments over carbon-supported Pd catalysts. The remaining solid residu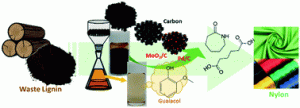 e during lignin extraction was also used as a carbon support to load Pd catalysts. The produced cyclohexanone and cyclohexanol were further converted to caprolactam and adipic acid, the main reagents used to produce nylon-6 and nylon-6,6 fibres, respectively. This process was demonstrated using real kraft and Klason lignin released from industry. The mass of guaiacol produced from 20 g of kraft lignin was 0.07 g, yielding either 0.032 g caprolactam or 0.043 g adipic acid, with a total yield of 0.15–0.25%. This study sheds light on utilizing waste lignin as a resource by producing not only guaiacol raw material for the production of high value-added nylon, but also a carbon support used for catalytic conversion.
e during lignin extraction was also used as a carbon support to load Pd catalysts. The produced cyclohexanone and cyclohexanol were further converted to caprolactam and adipic acid, the main reagents used to produce nylon-6 and nylon-6,6 fibres, respectively. This process was demonstrated using real kraft and Klason lignin released from industry. The mass of guaiacol produced from 20 g of kraft lignin was 0.07 g, yielding either 0.032 g caprolactam or 0.043 g adipic acid, with a total yield of 0.15–0.25%. This study sheds light on utilizing waste lignin as a resource by producing not only guaiacol raw material for the production of high value-added nylon, but also a carbon support used for catalytic conversion.
87. Boosting Thermal Stability of Volatile Os Catalysts by Downsizing to Atomically Dispersed Species
Journal paperOs-based catalysts present remarkable catalytic activity; however, their use has been limited by the undesirable side reactions that generate hi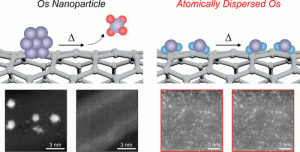 ghly toxic and volatile OsO4 even at room temperature. Herein, we demonstrate that the thermal stability of Os-based catalysts can be dramatically improved by downsizing Os nanoparticles (NPs) into atomically dispersed species. We observed that Os NPs were converted into OsO4 after calcination at 250 °C followed by sublimation, whereas single Os sites retained their structure after calcination. Temperature-programmed oxidation analysis confirmed that Os NPs started to undergo oxidation at 130 °C, whereas atomically dispersed Os preserved its state up to 300 °C. The CO oxidation activity of the atomically dispersed Os catalyst at 400 °C (100% conversion) was stably preserved over 30 h. By contrast, the activity of Os NP catalyst declined drastically. This study highlights the unique catalytic behavior of atomically dispersed catalysts, which is distinct from that of NP-based catalysts.
ghly toxic and volatile OsO4 even at room temperature. Herein, we demonstrate that the thermal stability of Os-based catalysts can be dramatically improved by downsizing Os nanoparticles (NPs) into atomically dispersed species. We observed that Os NPs were converted into OsO4 after calcination at 250 °C followed by sublimation, whereas single Os sites retained their structure after calcination. Temperature-programmed oxidation analysis confirmed that Os NPs started to undergo oxidation at 130 °C, whereas atomically dispersed Os preserved its state up to 300 °C. The CO oxidation activity of the atomically dispersed Os catalyst at 400 °C (100% conversion) was stably preserved over 30 h. By contrast, the activity of Os NP catalyst declined drastically. This study highlights the unique catalytic behavior of atomically dispersed catalysts, which is distinct from that of NP-based catalysts.
86. Boosting support reducibility and metal dispersion by exposed surface atom control for highly active supported metal catalysts
Journal paperFor oxide supported metal catalysts, support reducibility and metal dispersion are the key factors to determine the activity and selectivity in many essential reactions involving redox process. Herein, we tuned the exposed surface atoms of the catalyst by facet control and doping methods, w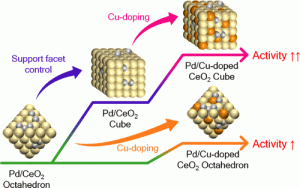 hich were simultaneously applied to boost the reducibility and metal dispersion of an oxide support. Pd supported on Cu-doped CeO2 (Pd/CDC) for water-gas shift reaction (WGSR) was considered a model system; Cu was doped into the cubes and octahedra CeO2 enclosed with (100) and (111) facets, respectively. By systematic combination of DFT calculations and experimental analyses, the WGSR activity of the Pd/CDC cube was verified to synergistically increase by more than just the sum of the morphology and Cu doping effects. The effect of each tuning method on the activity was further investigated from a mechanistic perspective. This work presents a rational design knowledge to enhance catalytic activity that can be extended to a wide range of supported metal systems.
hich were simultaneously applied to boost the reducibility and metal dispersion of an oxide support. Pd supported on Cu-doped CeO2 (Pd/CDC) for water-gas shift reaction (WGSR) was considered a model system; Cu was doped into the cubes and octahedra CeO2 enclosed with (100) and (111) facets, respectively. By systematic combination of DFT calculations and experimental analyses, the WGSR activity of the Pd/CDC cube was verified to synergistically increase by more than just the sum of the morphology and Cu doping effects. The effect of each tuning method on the activity was further investigated from a mechanistic perspective. This work presents a rational design knowledge to enhance catalytic activity that can be extended to a wide range of supported metal systems.
85. Carbothermal Shock-Induced Bifunctional Pt-Co Alloy Electrocatalysts for High-Performance Seawater Batteries
Journal paperSeawater batteries consisting of Na anode, Na super-ionic conductor separators, and seawater catholytes have received wide attention because o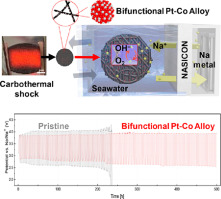 f their theoretical specific capacity of 1160 mAh g−1 and cost-effective Na anode in comparison to rare-earth Li. However, large overpotential during charge and discharge caused by parasitic reactions limits their practical applications. In this work, we employ the bifunctional Pt-Co alloy electrocatalysts produced by carbothermal shock (CTS) method to improve the oxygen evolution and reduction reaction activities of seawater batteries. The CTS induced Pt-Co alloy nanoparticles are well synthesized and dispersed on a carbon current collector within a few s, resulting in improved overpotential and cycle endurance of seawater batteries compared to pristine carbon cathode. In particular, the cell can operate for over 500 h in a seawater catholyte at a fixed capacity of 0.25 mA cm−2 without significant performance degradation. Furthermore, CTS can be readily applied to large-area prismatic seawater battery cells. We observe excellent cyclability in a large-scale seawater battery, suggesting that bifunctional Pt-Co alloy electrocatalysts produced by CTS are viable for use in seawater batteries.
f their theoretical specific capacity of 1160 mAh g−1 and cost-effective Na anode in comparison to rare-earth Li. However, large overpotential during charge and discharge caused by parasitic reactions limits their practical applications. In this work, we employ the bifunctional Pt-Co alloy electrocatalysts produced by carbothermal shock (CTS) method to improve the oxygen evolution and reduction reaction activities of seawater batteries. The CTS induced Pt-Co alloy nanoparticles are well synthesized and dispersed on a carbon current collector within a few s, resulting in improved overpotential and cycle endurance of seawater batteries compared to pristine carbon cathode. In particular, the cell can operate for over 500 h in a seawater catholyte at a fixed capacity of 0.25 mA cm−2 without significant performance degradation. Furthermore, CTS can be readily applied to large-area prismatic seawater battery cells. We observe excellent cyclability in a large-scale seawater battery, suggesting that bifunctional Pt-Co alloy electrocatalysts produced by CTS are viable for use in seawater batteries.
84. Influence of Pt Size and CeO2 Morphology at the Pt-CeO2 Interface in CO Oxidation
Journal paperUnderstanding the inherent catalytic nature of the interface between metal nanoparticles (NPs) and oxide supports enables the rational design of metal-support interactions for high catalytic performance. Electronic interactions at the metal-oxide interface create active interfacial sites that produce distinctive catalytic functions. However, because the overall catalytic properties of the interface are influenced by several complex structural factors, it is difficult to express the catalytic activity induced by the interfacial site through a simple description. Based on a combinatorial stud of density functional theory calculations and catalytic experiments, we focus on two structural design factors of metal NP-supported oxide catalysts: the size of Pt NPs and the morphology of the CeO2 support. Pt NPs with a size of 1, 2, and 3 nm were supported on the surface of CeO2-cube ({100} facet) and -octahedron ({111} facet) nanocrystals. During catalytic CO oxidation, the activity of Pt/CeO2- cube was higher than that of Pt/CeO2-octahedron, regardless of the size of NPs. Although 1 nm Pt NPs donate a similar number of electrons per Pt atom to CeO2-cubes and CeO2-octahedra, the inherently low oxygen va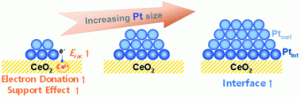 cancy formation energy of the CeO2(100) surface leads to the higher catalytic activity of the Pt-CeO2-cube interface. However, the intrinsic catalytic activity of the interface between Pt NPs and two CeO2 nanocrystals converges as the size of Pt NPs increases. Because large Pt NPs interact more strongly with CeO2(100) than CeO2(111), the positive effect of the low vacancy formation energy of CeO2(100) is compensated by the strengthened Pt-O interaction. This study elucidates how the interfaces formed between the shape-controlled CeO2 and the size-controlled Pt NPs affect the resultant catalytic activity.
cancy formation energy of the CeO2(100) surface leads to the higher catalytic activity of the Pt-CeO2-cube interface. However, the intrinsic catalytic activity of the interface between Pt NPs and two CeO2 nanocrystals converges as the size of Pt NPs increases. Because large Pt NPs interact more strongly with CeO2(100) than CeO2(111), the positive effect of the low vacancy formation energy of CeO2(100) is compensated by the strengthened Pt-O interaction. This study elucidates how the interfaces formed between the shape-controlled CeO2 and the size-controlled Pt NPs affect the resultant catalytic activity.
83. Selective phase transformation of layered double hydroxides into mixed metal oxides for catalytic CO oxidation
Journal paperPhase transformation from layered double hydroxides (LDHs) into mixed metal oxides (MMOs) has been widely used in v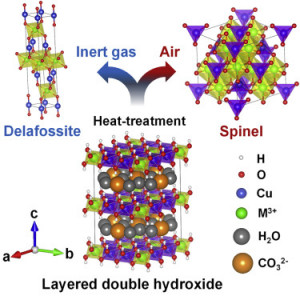 arious catalytic applications owing to its numerous advantages over conventional synthesis methods. Herein we report the results of selective phase transformation of LDHs into spinels and delafossites for the preparation of phase-pure MMO catalysts. Pure cuprous delafossites and cupric spinels were selectively obtained through heat treatment of Cu-based LDHs followed by post-treatments. This enabled the study of the crystalline-phase-dependent CO oxidation activity of the MMO catalysts and their physicochemical properties. The spinel catalysts exhibited higher CO oxidation activities, in comparison with those of the delafossites, with greater redox properties and improved active sites for CO adsorption. Although the crystalline phases were derived from the same LDH precursors, the catalytic properties of the end product were greatly influenced by their crystal structures.
arious catalytic applications owing to its numerous advantages over conventional synthesis methods. Herein we report the results of selective phase transformation of LDHs into spinels and delafossites for the preparation of phase-pure MMO catalysts. Pure cuprous delafossites and cupric spinels were selectively obtained through heat treatment of Cu-based LDHs followed by post-treatments. This enabled the study of the crystalline-phase-dependent CO oxidation activity of the MMO catalysts and their physicochemical properties. The spinel catalysts exhibited higher CO oxidation activities, in comparison with those of the delafossites, with greater redox properties and improved active sites for CO adsorption. Although the crystalline phases were derived from the same LDH precursors, the catalytic properties of the end product were greatly influenced by their crystal structures.
82. Layered Double Hydroxide-Derived Intermetallic Ni3GaC0.25 Catalysts for Dry Reforming of Methane
Journal paperA NiMgGa-layered double hydroxide (NMG-LDH) is synthesized as an efficient catalyst precursor for dry reforming of methane (DRM). NMG-LDH is converted to an intermetallic Ni3Ga/MgO catalyst upon reduction. Compared to a monometallic Ni/MgO catalyst prepared from NiMg-LDH, the Ni3Ga/MgO catalyst exhibits high CH4 (∼48%) and CO2 (∼52%) conversions as well as excellent stability against coking during DRM. T he reversible phase transition between intermetallic Ni3Ga and Ni3GaCx is demonstrated by in situ characterizations with the interstitial carbon being involved in the catalytic cycle of DRM to produce CO and H2. According to density functional theory calculations and the experimental study, the LDH-derived Ni3Ga intermetallic catalyst is converted to the Ni3GaC0.25 phase when carbon atoms dissociated from CH4 penetrate into the octahedral interstices of the Ni3Ga lattice during DRM at 600 °C. The formed Ni3GaC0.25 is proven effective in converting the interstitial carbon rapidly into CO to suppress its conversion to the coke, thus improving the stability of the catalyst.
he reversible phase transition between intermetallic Ni3Ga and Ni3GaCx is demonstrated by in situ characterizations with the interstitial carbon being involved in the catalytic cycle of DRM to produce CO and H2. According to density functional theory calculations and the experimental study, the LDH-derived Ni3Ga intermetallic catalyst is converted to the Ni3GaC0.25 phase when carbon atoms dissociated from CH4 penetrate into the octahedral interstices of the Ni3Ga lattice during DRM at 600 °C. The formed Ni3GaC0.25 is proven effective in converting the interstitial carbon rapidly into CO to suppress its conversion to the coke, thus improving the stability of the catalyst.
81. Methane oxidation to formaldehyde over vanadium oxide supported on various mesoporous silicas
Journal paperTo investigate the role of vanadium oxide supported on mesoporous silica (VOx/m-SiO2) catalysts in methane o xidation to formaldehyde, various catalysts were prepared. The type of m-SiO2 (SBA-15 and MCF-17), vanadium loading (1, 3, and 5%), and preparation method (wet impregnation; WI and dry impregnation; DI) were changed to produce VOx/m-SiO2 with different vanadium species. Because of the larger surface area and pore size, a higher dispersion of vanadium loading, 1% VOx/MCF-17(DI), showed the highest conversion (20.2%) in methane oxidation at 600 oC. Various characterizations revealed that DI was a better method to produce isolated tetrahedral monovanadate species in VOx/m-SiO2 catalysts than WI. As the vanadium loading was decreased from 5 to 1%, the methane conversion was further increased due to the higher degree of dispersion of monomeric VO4 generated in the catalysts with low vanadium loading. The combined results demonstrate that the dispersion of vanadium and the isolated monomeric VO4 phase increased when the vanadium catalyst was loaded on MCF-17 and prepared by the DI method.
xidation to formaldehyde, various catalysts were prepared. The type of m-SiO2 (SBA-15 and MCF-17), vanadium loading (1, 3, and 5%), and preparation method (wet impregnation; WI and dry impregnation; DI) were changed to produce VOx/m-SiO2 with different vanadium species. Because of the larger surface area and pore size, a higher dispersion of vanadium loading, 1% VOx/MCF-17(DI), showed the highest conversion (20.2%) in methane oxidation at 600 oC. Various characterizations revealed that DI was a better method to produce isolated tetrahedral monovanadate species in VOx/m-SiO2 catalysts than WI. As the vanadium loading was decreased from 5 to 1%, the methane conversion was further increased due to the higher degree of dispersion of monomeric VO4 generated in the catalysts with low vanadium loading. The combined results demonstrate that the dispersion of vanadium and the isolated monomeric VO4 phase increased when the vanadium catalyst was loaded on MCF-17 and prepared by the DI method.
80. Modified Metal–Organic Frameworks as Efficient Catalysts for Lignocellulosic Biomass Conversion
Journal paper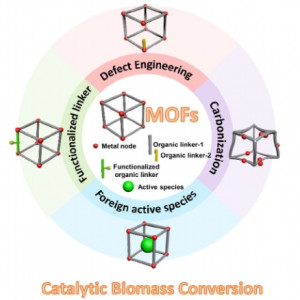 ating catalysts for lignocellulosic biomass conversion to valuable chemicals and fuels. Metal–organic frameworks (MOFs) are important catalysts because of their well-ordered porous structures and large surface areas. Although MOFs can be applied directly, four modification strategies can be used to alter their catalytic properties and improve catalyticperformance. In the first strategy, coordinatively unsaturated sites are created by changing the bonding state of the metal node. In the second approach, organic linkers with additional functional groups or active elements are used. In the third strategy, MOFs and other active elements are combined. In the final approach, MOFs are carbonized to produce carbon-supported metal catalysts. We review the applications of modified MOFs for the catalytic conversion of biomass derivatives and discuss the factors that contribute to their improved catalytic performance.
ating catalysts for lignocellulosic biomass conversion to valuable chemicals and fuels. Metal–organic frameworks (MOFs) are important catalysts because of their well-ordered porous structures and large surface areas. Although MOFs can be applied directly, four modification strategies can be used to alter their catalytic properties and improve catalyticperformance. In the first strategy, coordinatively unsaturated sites are created by changing the bonding state of the metal node. In the second approach, organic linkers with additional functional groups or active elements are used. In the third strategy, MOFs and other active elements are combined. In the final approach, MOFs are carbonized to produce carbon-supported metal catalysts. We review the applications of modified MOFs for the catalytic conversion of biomass derivatives and discuss the factors that contribute to their improved catalytic performance.79. Atomically alloyed Fe-Co catalyst derived from a N-coordinated Co single-atom structure for CO2 hydrogenation
Journal paperWe report a stable and efficient Fe–Co catalyst derived from N-coordinated Co single-atom carbon (FeK/Co–NC) for CO2 convers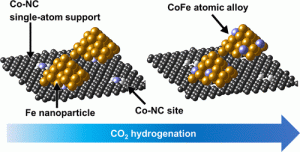 ion to long-chain hydrocarbons with a C5+ selectivity of up to 42.4% at a conversion of 51.7% at 300 °C and 2.5 MPa. Its performance remained stable over a time-on-stream of 100 h. The FeK/Co–NC catalyst exhibited less methane selectivity (21.6%) than the coimpregnated FeCoK/NC catalyst (33.8%), which is attributed to the Co–NC support, efficiently inducing Fe–Co alloy formation by atomically supplying Co into Fe nanoparticles. The Fe–Co alloy of the FeK/Co–NC catalyst remained stable in both carburized and oxide forms during the reaction. Density functional theory suggests that Fe–Co mixed oxides accelerate oxygen removal during the reverse water–gas shift, whereas Fe–Co mixed carbides promote chain growth to suppress methane formation during Fischer–Tropsch synthesis. Our combined experimental and theoretical study demonstrates the promoting effect of the Fe–Co atomic alloy structure for CO2 hydrogenation.
ion to long-chain hydrocarbons with a C5+ selectivity of up to 42.4% at a conversion of 51.7% at 300 °C and 2.5 MPa. Its performance remained stable over a time-on-stream of 100 h. The FeK/Co–NC catalyst exhibited less methane selectivity (21.6%) than the coimpregnated FeCoK/NC catalyst (33.8%), which is attributed to the Co–NC support, efficiently inducing Fe–Co alloy formation by atomically supplying Co into Fe nanoparticles. The Fe–Co alloy of the FeK/Co–NC catalyst remained stable in both carburized and oxide forms during the reaction. Density functional theory suggests that Fe–Co mixed oxides accelerate oxygen removal during the reverse water–gas shift, whereas Fe–Co mixed carbides promote chain growth to suppress methane formation during Fischer–Tropsch synthesis. Our combined experimental and theoretical study demonstrates the promoting effect of the Fe–Co atomic alloy structure for CO2 hydrogenation.
78. Revealing Charge Transfer at the Interface of Spinel Oxide and Ceria during CO Oxidation
Journal paperThe interface created between an active metal and an oxide support is known to affect the catalytic performance because of the charge transfer process. However, oxide–oxide interfaces produced by supported spinel oxide catalysts have been less studied owing to their complex interface structures and synthetic challenges. Herein, a synthetic strategy for Co3O4, Mn3O4, and Fe3O4 nanocubes (NCs) with a controlled CeO2 layer enables investigation of the role of the interface in catalytic oxidation. Notably, CeO2-deposited Co3O4 NCs exhibited a 12-times higher CO oxidation rate than the pristine Co3O4 NCs. In situ characterization demonstrates that the deposited CeO2 prevents the reduction of Co3O4 by supplying oxygen. The maximized interface resulting from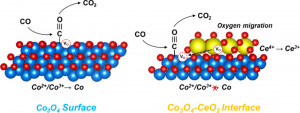 Co3O4 NCs with three facets covered by CeO2 layers was found to exhibit the highest CO oxidation rate even under O2-deficient conditions, which resulted from the versatile variation in the oxidation state. This study provides a comprehensive understanding of the Mars–van Krevelen mechanism occurring on the nanoscale at the Co3O4–CeO2 interfaces. The same activity trend and hot electron flow are observed for H2 oxidation reactions using catalytic nanodiodes, thereby demonstrating that the origin of the activity enhancement is charge transfer at the interface.
Co3O4 NCs with three facets covered by CeO2 layers was found to exhibit the highest CO oxidation rate even under O2-deficient conditions, which resulted from the versatile variation in the oxidation state. This study provides a comprehensive understanding of the Mars–van Krevelen mechanism occurring on the nanoscale at the Co3O4–CeO2 interfaces. The same activity trend and hot electron flow are observed for H2 oxidation reactions using catalytic nanodiodes, thereby demonstrating that the origin of the activity enhancement is charge transfer at the interface.
77. Al2O3-Coated Ni/CeO2 Nanoparticles as Coke-Resistant Catalyst for Dry Reforming of Methane
Journal paperNickel is considered an economically feasible catalyst for the dry reforming of methane (DRM) owing to its high activity. Because the highly endothermic DRM requires a high reaction temperature to activate both CH4 and CO2, deactivation of the Ni catalyst may be induced by sintering and carbon coking. To mitigate catalyst deactivation, Ni/CeO2 catalysts composed of monodisperse Ni nanoparticles supported on CeO2 nanorods are designed and coated with Al2O3 layers by atomic layer deposition (ALD). The performance of the catalyst in DRM and amount of carbon deposited are correlated with the thickness of the Al2O3 layer in the Ni/CeO2/Al2O3 catalysts. As the number of ALD cycles increases from 1 to 10, the conversion of CO2 and CH4 at 700 and 800 °C decreases, but the Ni/CeO2/Al2O3 catalysts remain coke-free as thermogravimetric analysis shows no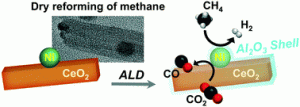 weight loss up to 800 °C. The Al2O3 layer generated by ALD curtails the coking substantially, but the weakly metallic character of Ni and blocking of Ni sites by the Al2O3 layer are the major factors contributing to decreasing the catalytic conversion. The ALD technique provides an efficient way to fabricate atomically controlled oxide layers for improving the stability of catalysts against coke deposition and sintering.
weight loss up to 800 °C. The Al2O3 layer generated by ALD curtails the coking substantially, but the weakly metallic character of Ni and blocking of Ni sites by the Al2O3 layer are the major factors contributing to decreasing the catalytic conversion. The ALD technique provides an efficient way to fabricate atomically controlled oxide layers for improving the stability of catalysts against coke deposition and sintering.
76. Structural Evolution of ZIF-67-Derived Catalysts for Furfural Hydrogenation
Journal paperZeolitic imidazolate framework-67 (ZIF-67) can be converted to metallic Co nanoparticles supported on N-doped carbon (Co/NC) through reduction. However, its unique properties, including extremely high surface area, isoreticular pore structure, and regular metal-organic network, disappear after high-temperature (>500 ℃) reduction. Aggregated CoOx particles reduce the number of surface-active sites, resulting in poor catalytic activity. If the original ZIF-67 structure is maintained after the high-temperature reduction, promoting the uniform distribution of active sites in the porous carbon, the catalytic performance can be further impro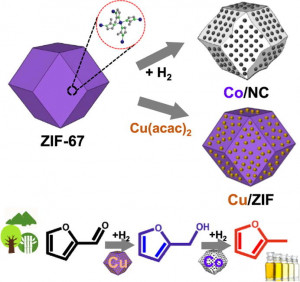 ved. Herein, the correlation between the catalytic furfural hydrogenation performance, Co/NC morphology, and oxidation state of Co was investigated as a function of the H2 reduction temperature and time. The reduction of ZIF-67 at 400 ℃ for 6 h yields a highly dispersed Co/NC catalyst, while preserving the overall morphology. The resulting Co/NC-400-6 catalyst exhibits the highest activity, promoting high selectivity toward 2-methylfuran. The product selectivity can be further altered by incorporating Cu into ZIF-67 to produce furfuryl alcohol. With proper H2 treatment to minimize the damage to the intrinsic surface area and pore structure, metal-organic frameworks can be utilized as high-performance heterogeneous catalysts by maximizing the distribution of active sites.
ved. Herein, the correlation between the catalytic furfural hydrogenation performance, Co/NC morphology, and oxidation state of Co was investigated as a function of the H2 reduction temperature and time. The reduction of ZIF-67 at 400 ℃ for 6 h yields a highly dispersed Co/NC catalyst, while preserving the overall morphology. The resulting Co/NC-400-6 catalyst exhibits the highest activity, promoting high selectivity toward 2-methylfuran. The product selectivity can be further altered by incorporating Cu into ZIF-67 to produce furfuryl alcohol. With proper H2 treatment to minimize the damage to the intrinsic surface area and pore structure, metal-organic frameworks can be utilized as high-performance heterogeneous catalysts by maximizing the distribution of active sites.
75. Cu2O(100) surface as an active site for catalytic furfural hydrogenation
Journal paperIn order to investigate the major active site of Cu-based catalysts in furfural (FAL) hydrogenation, theoretical
calculations were combined with empirical analyses. The adsorption of FAL and H2 on the Cu(111), CuO(100),
and Cu2O(100) surfaces was compared based on density functional theory (DFT) calculations. The migration
barrier of the dissociatively adsorbed H atoms on different surfaces was also calculated. It is demonstrated that
the Cu2O(100) surface has the largest FAL adsorption energy of 1.63 eV and an appropriate Cu‒Cu distance for
adsorption and preferential dissociation of the H2 molecule. To correlate the DFT results with catalytic experiments, mesoporous copper oxides (m-CuO) were prepared und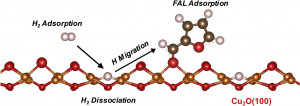 er controlled reduction conditions. The overall activity of the m-CuO catalysts is determined by the concentration of exposed Cu+. The combined results from DFT calculations and experiments show that Cu2O is a major active species promoting the high activity of FAL hydrogenation.
er controlled reduction conditions. The overall activity of the m-CuO catalysts is determined by the concentration of exposed Cu+. The combined results from DFT calculations and experiments show that Cu2O is a major active species promoting the high activity of FAL hydrogenation.
74. Recycling Carbon Dioxide through Catalytic Hydrogenation: Recent Key Developments and Perspectives
Journal paperRecycling CO2 as a renewable carbon source for the production of high-value fuels and chemicals has drawn global attention lately as a promising method to mitigate climate change and lessen dependence on fossil fuels. Among the available CO2-recycling options, catalytic CO2 hydrogenation is the most realistic and attractive choice if the hydrogen is produced using a renewable energy source. Depending on the nature of the catalyst, CO2 hydrogenation has distinct reaction pat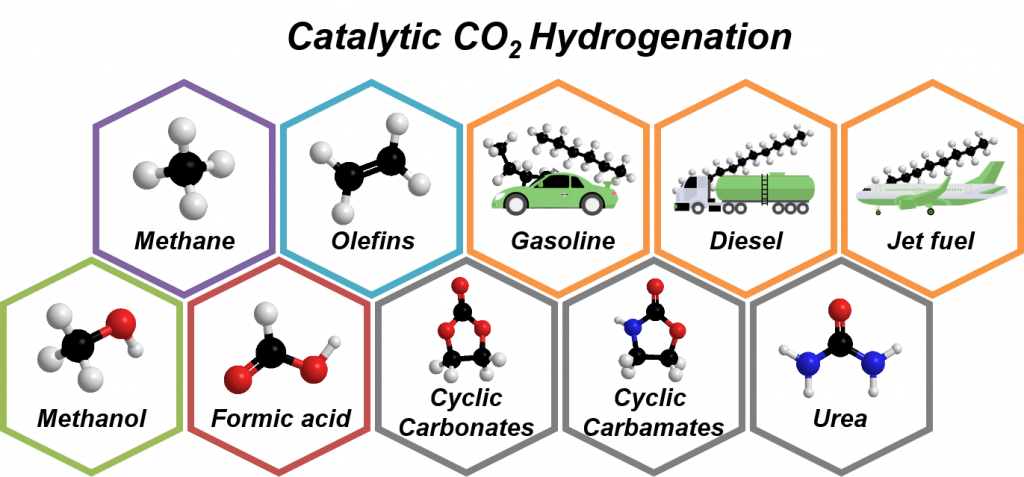 hways, and various value-added hydrocarbons can be produced. Intense research has recently developed high-performance catalysts, identified clear reaction pathways, and deepened the understanding of the reaction mechanisms. In this review, we present an overview of recent key advances in catalytic CO2 hydrogenation to high-value hydrocarbons and oxygenates that have large market sizes, such as formic acid, methanol, methane, and light olefins, as well as liquid fuels, in terms of the catalyst design, catalytic performance, and reaction mechanism. In addition, the current technical challenges and perspectives on CO2 conversion processes are discussed with regard to climate change mitigation.
hways, and various value-added hydrocarbons can be produced. Intense research has recently developed high-performance catalysts, identified clear reaction pathways, and deepened the understanding of the reaction mechanisms. In this review, we present an overview of recent key advances in catalytic CO2 hydrogenation to high-value hydrocarbons and oxygenates that have large market sizes, such as formic acid, methanol, methane, and light olefins, as well as liquid fuels, in terms of the catalyst design, catalytic performance, and reaction mechanism. In addition, the current technical challenges and perspectives on CO2 conversion processes are discussed with regard to climate change mitigation.
73. Cobalt Ferrite Nanoparticles to Form a Catalytic Co–Fe Alloy Carbide Phase for Selective CO2 Hydrogenation to Light Olefins
Journal paperMonodisperse nanoparticles (NPs) of CoFe2O4 were synthesized as efficient catalyst precursors for CO2 hydrogenation to produce high value-added C2–C4 olefin products, which are important building blocks for the chemical industry. The resulting Na-promoted CoFe2O4 catalysts supported on carbon nanotubes (Na–CoFe2O4/CNT) exhibited high CO2 conversion (∼34%) and light olefin selectivity (∼39%), outperforming other reported Fe-based catalysts under similar reaction conditions. Their performance was superior to that of single-metal NP catalysts (Na–Fe3O4/CNT and Na–Co/CNT) and a physically mixed (Na–Fe3O4 + Co)/CNT catalyst. The superior performance of the Na–CoFe2O4/CNT catalyst can be attributed to the facile formation of a unique bimetallic alloy carbide (Fe1–xCox)5C2, which results in higher CO2 conversion and better selectivity toward light ol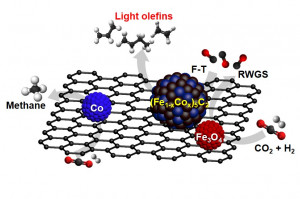 efins in comparison with conventional χ-Fe5C2 active sites derived from Fe-only catalysts and significantly improved heavy hydrocarbon (C2+) formation in comparison with the Co2C sites of Co-only catalysts. The single-source precursor CoFe2O4 exclusively forms a single-phase alloy carbide promoted by the Na promoter, whereas the mixed (Na–Fe3O4 + Co) precursor forms an isolated Co phase with the alloy carbide phase, promoting undesirable CH4 formation. An optimal value of x ≤ 0.2 for (Fe1–xCox)5C2 was predicted using the cluster expansion method and density functional theory, resulting in a stable bimetallic alloy structure.
efins in comparison with conventional χ-Fe5C2 active sites derived from Fe-only catalysts and significantly improved heavy hydrocarbon (C2+) formation in comparison with the Co2C sites of Co-only catalysts. The single-source precursor CoFe2O4 exclusively forms a single-phase alloy carbide promoted by the Na promoter, whereas the mixed (Na–Fe3O4 + Co) precursor forms an isolated Co phase with the alloy carbide phase, promoting undesirable CH4 formation. An optimal value of x ≤ 0.2 for (Fe1–xCox)5C2 was predicted using the cluster expansion method and density functional theory, resulting in a stable bimetallic alloy structure.
72. Interfacial Effect of Pd supported on Mesoporous Oxide for Catalytic Furfural Hydrogenation
Journal paperIn heterogeneous catalysis, it is of utmost importance to minimize the consumption of noble metals for commercialization. One of the strategies is to use oxide supports, which enhance the catalytic activity, while maintaining a high dispersion of noble metals on themselves. Here, we investigate the enhancement of catalytic properties of Pd on different types of mesoporous oxide supports in liquid-phase furfural (FAL) hydrogenation. Ordered mesoporous Co3O4, MnO2, NiO, CeO2, and Fe2O3 are prepared by the nanocasting and highly dispersed Pd nanoparticle catalyst on mesoporous oxides are obtained by the chemical reduction method. It is revealed that mesoporous oxides play an important role on Pd dispersion as well as the redox behavi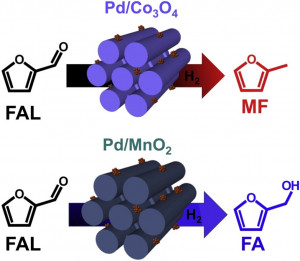 or of Pd, which determines the final FAL conversion. Among the catalysts used, Pd/Co3O4 shows the highest conversion in FAL hydrogenation and distinct product selectivity toward 2-methylfuran (MF). While FAL is converted via two distinct pathways to produce either furfuryl alcohol (FA) via aldehyde hydrogenation or MF via hydrogenolysis, MF as a secondary product is derived from FA via the hydrogenolysis of C‒O over the Pd/Co3O4 catalyst. It is revealed that FAL is hydrogenated to FA preferentially on the Pd surface; then the secondary hydrogenolysis to MF from FA is further promoted at the interface between Pd and Co3O4. We confirm that the reaction pathway over Pd/Co3O4 is totally different from other catalysts such as Pd/MnO2, which produces FA dominantly. The characteristics of the mesoporous oxides influence the Pd-oxide interfaces, which determine the activity and selectivity in FAL hydrogenation.
or of Pd, which determines the final FAL conversion. Among the catalysts used, Pd/Co3O4 shows the highest conversion in FAL hydrogenation and distinct product selectivity toward 2-methylfuran (MF). While FAL is converted via two distinct pathways to produce either furfuryl alcohol (FA) via aldehyde hydrogenation or MF via hydrogenolysis, MF as a secondary product is derived from FA via the hydrogenolysis of C‒O over the Pd/Co3O4 catalyst. It is revealed that FAL is hydrogenated to FA preferentially on the Pd surface; then the secondary hydrogenolysis to MF from FA is further promoted at the interface between Pd and Co3O4. We confirm that the reaction pathway over Pd/Co3O4 is totally different from other catalysts such as Pd/MnO2, which produces FA dominantly. The characteristics of the mesoporous oxides influence the Pd-oxide interfaces, which determine the activity and selectivity in FAL hydrogenation.
71. Efficient Hydrogenation Catalytic Model Hosted in Stable Hyper-crosslinked Porous-Organic-Polymer: From Fatty Acids to Bio-based Alkanes Diesel Synthesis
Journal paperIn this study, Pd-based catalytic model hosted over nitrogen enriched fibrous Porous-Organic-Polymer (POP) is established to execute hydrodeoxygenation of various vegetable oils in producing potential large-scale renewable diesel. Here we report a cost effective synthesis strategy of a new microporous hypercrosslinked POP through the FeCl3 assisted Friedel-Crafts alkylation reaction, followed by fabrication of Pd0-NPs (2-3 nm) with solid gas phase hydrogenation route to deliver a novel catalytic system. This catalyst (called Pd@PPN) exhibits versatile catalytic performance for different types of vegetable oils including palm oil, soybean oil, sunflower oil and rapeseed oil to furnish long chain diesel range alkanes. The catalyst is comprehensively characterized by various spectroscopic tools and shows high stability during five runs of recycling without the leaching of Pd occuring. Our results further reveal that direct decarbonylation (DCN) pathway of fatty acid to produce alkanes with one carbon less is the dominated mechanism. At optimized conditions, using stearic acid to represent the long linear carboxylic acids in the vegetable oils, up to 90 % conversion with 83 % selectivity of C17-alkane has been achieved on our fabricated catalyst. Density functional theory (DFT) calculations are performed to provide insights into the electronic properties of the catalyst, the mechanistic reaction pathway, the crucial role of catalyst surface and the product selectivity trend. The strong interaction between corrugated polymer-frame-structure and the Pd-NPs suggests there is the presence of high density step sites on the fabricated Pd-NP anchored within the cage of polymer structure. DFT calcualtions also reveal the strong promotional effect of step sites and charge transfer in facilitating rate-limiting steps during the decarbonylation (DCN) pathway and removal of strongly bound intermediates formed during the process, therefore explain the high activity of the fabricated Pd@PPN catayst for the hydrodeoxygenation (HDO) conversion to produce bio-based alkanes diesel.
70. Synergistic effect of quinary molten salts and ruthenium catalyst for high-power-density lithium-carbon dioxide cell
Journal paperWit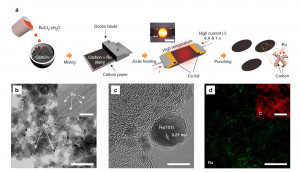 h a recent increase in interest in metal-gas batteries, the lithium-carbon dioxide cell has attracted considerable attention because of its extraordinary carbon dioxide-capture ability during the discharge process and its potential application as a power source for Mars exploration. However, owing to the stable lithium carbonate discharge product, the cell enables operation only at low current densities, which significantly limits the application of lithium-carbon dioxide batteries and effective carbon dioxide-capture cells. Here, we investigate a high-performance lithium-carbon dioxide cell using a quinary molten salt electrolyte and ruthenium nanoparticles on the carbon cathode. The nitrate-based molten salt electrolyte allows us to observe the enhanced carbon dioxide-capture rate and the reduced dischargecharge over-potential gap with that of conventional lithium-carbon dioxide cells. Furthermore, owing to the ruthernium catalyst, the cell sustains its performance over more than 300 cycles at a current density of 10.0 A g−1 and exhibits a peak power density of 33.4mWcm−2.
h a recent increase in interest in metal-gas batteries, the lithium-carbon dioxide cell has attracted considerable attention because of its extraordinary carbon dioxide-capture ability during the discharge process and its potential application as a power source for Mars exploration. However, owing to the stable lithium carbonate discharge product, the cell enables operation only at low current densities, which significantly limits the application of lithium-carbon dioxide batteries and effective carbon dioxide-capture cells. Here, we investigate a high-performance lithium-carbon dioxide cell using a quinary molten salt electrolyte and ruthenium nanoparticles on the carbon cathode. The nitrate-based molten salt electrolyte allows us to observe the enhanced carbon dioxide-capture rate and the reduced dischargecharge over-potential gap with that of conventional lithium-carbon dioxide cells. Furthermore, owing to the ruthernium catalyst, the cell sustains its performance over more than 300 cycles at a current density of 10.0 A g−1 and exhibits a peak power density of 33.4mWcm−2.
69. Structure-Dependent Catalytic Properties of Mesoporous Cobalt Oxides in Furfural Hydrogenation.
Journal paper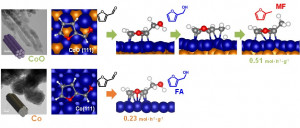 As the development of noble metal free catalysts became important in the biomass conversion, catalytic hydrogenation of furfural (FAL) is investigated over ordered mesoporous cobalt oxide (m-Co3O4). When m-Co3O4 is reduced at 350 and 500 °C in hydrogen, the original crystal structure of Co3O4 is changed to CoO and Co, respectively. Here we examine the effect of the structure, porosity, and oxidation state of m-Co3O4 to identify catalytically active species for hydrogenation of FAL. Among cobalt oxide catalysts having different crystal structures and symmetry, m-CoO having p6mm symmetry exhibits the highest activity. In product selectivity, the CoO phase induces FAL hydrogenolysis by selective production of 2-methyl furan (MF), while the Co3O4 and Co phases promote preferential hydrogenation of side chain (carbonyl group) of FAL to furfuryl alcohol. Density functional theory calculations also reveal that the adsorption of FAL on CoO(111) is higher than Co(111). Overall, these studies demonstrate that CoO as the most active phase is responsible for the high FAL conversion and the distinct pathway of FAL to MF.
As the development of noble metal free catalysts became important in the biomass conversion, catalytic hydrogenation of furfural (FAL) is investigated over ordered mesoporous cobalt oxide (m-Co3O4). When m-Co3O4 is reduced at 350 and 500 °C in hydrogen, the original crystal structure of Co3O4 is changed to CoO and Co, respectively. Here we examine the effect of the structure, porosity, and oxidation state of m-Co3O4 to identify catalytically active species for hydrogenation of FAL. Among cobalt oxide catalysts having different crystal structures and symmetry, m-CoO having p6mm symmetry exhibits the highest activity. In product selectivity, the CoO phase induces FAL hydrogenolysis by selective production of 2-methyl furan (MF), while the Co3O4 and Co phases promote preferential hydrogenation of side chain (carbonyl group) of FAL to furfuryl alcohol. Density functional theory calculations also reveal that the adsorption of FAL on CoO(111) is higher than Co(111). Overall, these studies demonstrate that CoO as the most active phase is responsible for the high FAL conversion and the distinct pathway of FAL to MF.
68. Highly Dispersed Pd Catalysts Supported on Various Carbons for Furfural Hydrogenation
Journal paperFurfural (FAL), on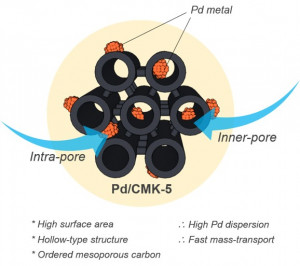 e of the important platform molecules derived from lignocellulosic biomass, can be converted into valuable chemicals such as furfuryl alcohol or cyclopentanone via hydrogenation. While carbon materials have been used as versatile catalyst supports for FAL hydrogenation, systematic studies on the structure of the catalytic performances are lacking. In this work, we prepare various types of carbon supports to investigate the impact of carbon structures for Pd-catalyzed FAL hydrogenation. Mesoporous carbons, including CMK-3, CMK-5, CMK-8, and MSU-F-C, as well as carbon nanotube and Vulcan XC are used as carbon supports. For the preparation of highly dispersed Pd-supported carbon (Pd/C) catalysts, chemical reduction by sodium borohydride is applied, in which trisodium citrate plays a critical role in anchoring small Pd clusters on the carbons. In the liquid-phase hydrogenation of FAL, CMK-5 with the largest surface area and hexagonal hollow tubular framework is proven to be the most efficient carbon support for Pd/C catalysts, with the highest conversion of FAL in both 2-propanol (100%) and water (86.4%) solvents. It is also demonstrated that the product selectivity in FAL hydrogenation over various Pd/C catalysts is changed dramatically depending on the type of solvent. The Pd/C catalysts exhibit similar fractions of product distributions containing furfuryl alcohol, cyclopentanol, tetrahydrofurfuryl alcohol, and minor products in 2-propanol. However, the production of cyclopentanone is increased when water is used as a solvent.
e of the important platform molecules derived from lignocellulosic biomass, can be converted into valuable chemicals such as furfuryl alcohol or cyclopentanone via hydrogenation. While carbon materials have been used as versatile catalyst supports for FAL hydrogenation, systematic studies on the structure of the catalytic performances are lacking. In this work, we prepare various types of carbon supports to investigate the impact of carbon structures for Pd-catalyzed FAL hydrogenation. Mesoporous carbons, including CMK-3, CMK-5, CMK-8, and MSU-F-C, as well as carbon nanotube and Vulcan XC are used as carbon supports. For the preparation of highly dispersed Pd-supported carbon (Pd/C) catalysts, chemical reduction by sodium borohydride is applied, in which trisodium citrate plays a critical role in anchoring small Pd clusters on the carbons. In the liquid-phase hydrogenation of FAL, CMK-5 with the largest surface area and hexagonal hollow tubular framework is proven to be the most efficient carbon support for Pd/C catalysts, with the highest conversion of FAL in both 2-propanol (100%) and water (86.4%) solvents. It is also demonstrated that the product selectivity in FAL hydrogenation over various Pd/C catalysts is changed dramatically depending on the type of solvent. The Pd/C catalysts exhibit similar fractions of product distributions containing furfuryl alcohol, cyclopentanol, tetrahydrofurfuryl alcohol, and minor products in 2-propanol. However, the production of cyclopentanone is increased when water is used as a solvent.
67. Integration of Interfacial and Alloy Effects to Modulate Catalytic Performance of MOF derived Cu-Pd Nanocrystals toward Hydrogenolysis of 5-hydroxymethylfurfural
Journal paper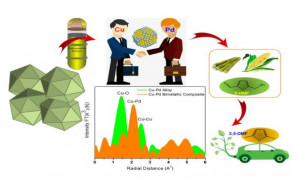 Selective formation of 2,5-dimethylfuran (DMF) by hydrogenolysis of lignocellulosic biomass derived 5-hydroxymethylfurfural (HMF) is highly desirable for renewable liquid biofuel production. Here we synthesised CuPd bimetallic nanoparticles embedded in carbon matrix (CuPd@C) by simple pyrolysis of Pd impregnated Cu-based metal-organicframeworks (MOFs) followed by conventional hydrogenation route. It was found that CuPd@C-B (solid-gas phase hydrogenation route) with Cu-Pd bimetallic alloying exhibited brilliant catalytic performance at 120ºC under 15 bar H2 pressure to produce liquid DMF biofuel with 96.5% yield from HMF as compared with the Cu-Pd@C-A catalyst (liquid phase hydrogenation route) of 46.4% yield under the same condition. XPS and XANES studies reveals that Pd in Cu-Pd@C-B catalyst is electronically promoted by Cu with the unique intrinsic synergy of increased Pd-Pd bond distance and decreased Cu-Cu bond length, which eventually modulate the local atomic structural environment and result in enhanced catalytic activity. Moreover, the entrapped bimetallic nanoparticles with carbon shells in Cu-Pd@C-B catalyst further protect the active catalytic site from migration, aggregation, and leaching during hydrogenolysis reaction and improve the stability of the catalyst.
Selective formation of 2,5-dimethylfuran (DMF) by hydrogenolysis of lignocellulosic biomass derived 5-hydroxymethylfurfural (HMF) is highly desirable for renewable liquid biofuel production. Here we synthesised CuPd bimetallic nanoparticles embedded in carbon matrix (CuPd@C) by simple pyrolysis of Pd impregnated Cu-based metal-organicframeworks (MOFs) followed by conventional hydrogenation route. It was found that CuPd@C-B (solid-gas phase hydrogenation route) with Cu-Pd bimetallic alloying exhibited brilliant catalytic performance at 120ºC under 15 bar H2 pressure to produce liquid DMF biofuel with 96.5% yield from HMF as compared with the Cu-Pd@C-A catalyst (liquid phase hydrogenation route) of 46.4% yield under the same condition. XPS and XANES studies reveals that Pd in Cu-Pd@C-B catalyst is electronically promoted by Cu with the unique intrinsic synergy of increased Pd-Pd bond distance and decreased Cu-Cu bond length, which eventually modulate the local atomic structural environment and result in enhanced catalytic activity. Moreover, the entrapped bimetallic nanoparticles with carbon shells in Cu-Pd@C-B catalyst further protect the active catalytic site from migration, aggregation, and leaching during hydrogenolysis reaction and improve the stability of the catalyst.
66. Mesoporous mixed CuCo oxides as robust catalysts for liquid-phase furfural hydrogenation
Journal paperA series of highly ordered mesoporous CuCo oxide catalysts with a controlled composition are
successfull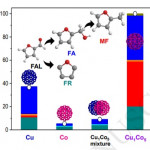 y synthesized by nanocasting from mesoporous silica, KIT-6 template. Liquidphase
y synthesized by nanocasting from mesoporous silica, KIT-6 template. Liquidphase
furfural (FAL) hydrogenation is carried out to find the optimal composition of the CuCo
oxide catalysts to achieve the best catalytic performance. As-prepared mesoporous mixed
CuCo oxides exhibit a high surface area (60‒135 m2‧ g-1) and a well-defined ordered
mesostructure with homogenous dispersion of Cu and Co. Among various compositions of CuxCoy
oxides (x = 1‒9) studied, the Cu1Co5 oxide catalyst shows the highest conversion in the hydrogenation of FAL, which is superior to those achieved with mesoporous monometallic oxides,
CuO and Co3O4. While 2-methylfuran is produced from furfuryl alcohol via aldehyde
hydrogenation and subsequent hydrogenolysis, the formation of 2-methylfuran increased with a
decrease in the Cu/Co ratio of the CuCo oxide catalyst. The mixed CuCo oxide catalyst is readily
reduced under the reaction environment to produce metallic CuCo as the active species. The
synergistic interactions between Cu and Co in the mixed CuCo oxide catalysts play an important
role in the outstanding catalytic performance for FAL hydrogenation, which could not be achieved
with either of the monometallic catalysts or their physical mixtures. The excellent stability and
recyclability of mesoporous mixed CuCo oxide catalysts as well as the exceptionally high
activity, surpassing those of the monometallic oxides, render them promising as a low-cost and
efficient catalyst for the industrial upgrading of biomass-derived FAL.
65. Enhanced hot electron generation by inverse metal–oxide interfaces on catalytic nanodiode
Journal paper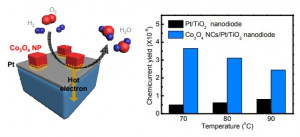 Identifying the electronic behavior of metal–oxide interfaces is essential for understanding the origin of catalytic properties and for engineering catalyst structures with the desired reactivity. For a mechanistic understanding of hot electron dynamics at inverse oxide/metal interfaces, we employed a new catalytic nanodiode by combining Co3O4 nanocubes (NCs) with a Pt/TiO2 nanodiode that exhibits nanoscale metal–oxide interfaces. We show that the chemicurrent, which is well correlated with the catalytic activity, is enhanced at the inverse oxide/metal (CoO/Pt) interfaces during H2 oxidation. Based on quantitative visualization of the electronic transfer efficiency with chemicurrent yield, we show that electronic perturbation of oxide/metal interfacial sites not only promotes the generation of hot electrons, but improves catalytic activity.
Identifying the electronic behavior of metal–oxide interfaces is essential for understanding the origin of catalytic properties and for engineering catalyst structures with the desired reactivity. For a mechanistic understanding of hot electron dynamics at inverse oxide/metal interfaces, we employed a new catalytic nanodiode by combining Co3O4 nanocubes (NCs) with a Pt/TiO2 nanodiode that exhibits nanoscale metal–oxide interfaces. We show that the chemicurrent, which is well correlated with the catalytic activity, is enhanced at the inverse oxide/metal (CoO/Pt) interfaces during H2 oxidation. Based on quantitative visualization of the electronic transfer efficiency with chemicurrent yield, we show that electronic perturbation of oxide/metal interfacial sites not only promotes the generation of hot electrons, but improves catalytic activity.
64. Catalytic CO oxidation over Au nanoparticles supported on CeO2 nanocrystals: Effect of the Au-CeO2 interface
Journal paper
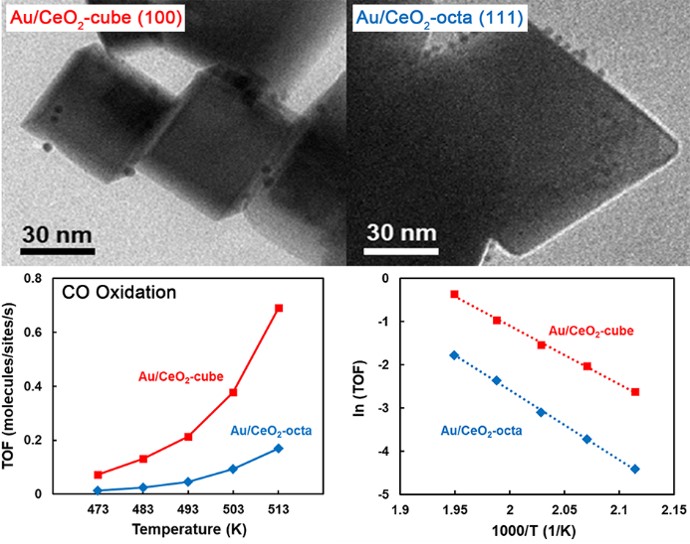
Gold nanoparticles (NPs) have attracted attention due to their superior catalytic performance in CO oxidation at low temperatures. Along with the size and shape of Au NPs, the catalytic function of Au-catalyzed CO oxidation can be further optimized by controlling the physicochemical properties of oxide-supporting materials. We applied a combinatorial approach of experimental analyses and theoretical interpretations to study the effect of a surface structure of supporting oxides and the corresponding CO oxidation activity of supported Au NPs. We synthesized Au NPs (average d ≈ 3 nm) supported on shape-controlled CeO2 nanocrystals, Au/CeO2 cubes and Au/CeO2 octahedra for experimental analyses. The catalysts were modeled as Au/CeO2(100) and Au/CeO2(111) via density functional theory (DFT) calculations. The DFT calculations showed that the O-C-O type reaction intermediate could be spontaneously formed at the Au-CeO2(100) interface upon sequential multi-CO adsorption, accelerating CO oxidation via the Mars-van Krevelen mechanism. The additional kinetic process required for O-C-O formation at the Au-CeO2(111) interface slowed down the reaction. The experimental turnover frequency (TOF) of the Au/CeO2 cubes was 4 times greater than that of the Au/CeO2 octahedra (under 0.05 bar CO and 0.13 bar O2). The increasing TOF as a function of CO partial pressure and the positive correlation between the reducibility of CeO2 and the catalytic activity of Au/CeO2 catalysts confirmed the theoretical prediction that CO molecules occupy the surface of Au NPs and that the oxidation of Au-bound CO occurs at the Au-CeO2 interface. Through a comparative study of DFT calculations and in-depth experimental analyses, we provide insights into the catalytic function of CeO2-supported Au NPs towards CO oxidation depending on the shape of CeO2 and ratio of CO/O2.
63. SiO2@V2O5@Al2O3 Core-Shell Catalysts with High Activity and Stability for Methane Oxidation to Formaldehyde
Journal paper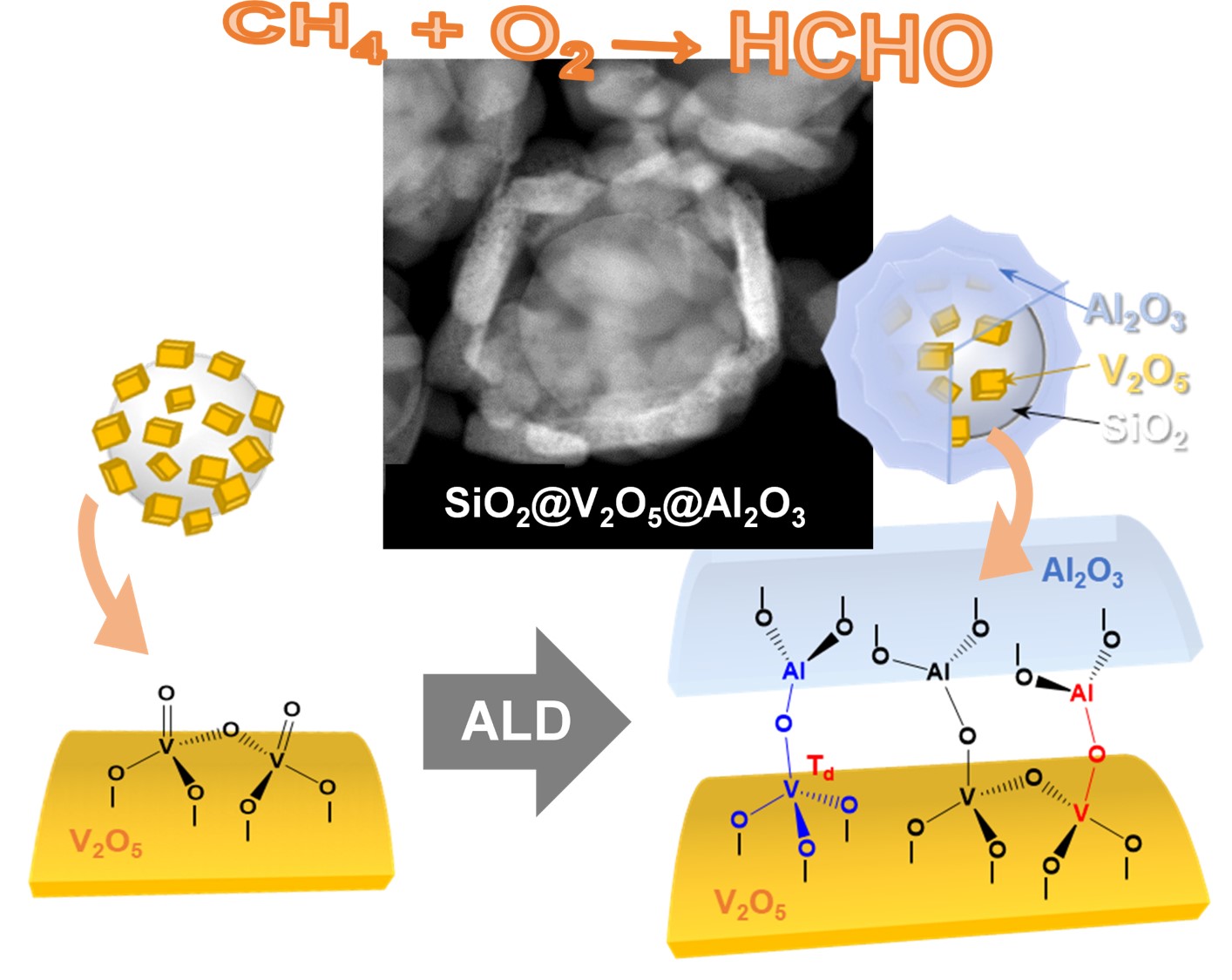 The stable tetrahedral geometry and high C-H bond dissociation energy of methane complicate its direct catalytic conversion; for example, the selective oxidation of methane to formaldehyde, which avoids the production of carbon dioxide by full oxidation and is therefore important for the versatile utilization of natural gas, is still viewed as challenging. Here, we utilize hydrothermal synthesis followed by atomic layer deposition (ALD) to prepare an efficient and thermally stable catalyst based on novel SiO2@V2O5@ Al2O3 core@shell nanostructures, showing that the thickness of Al2O3 shells over SiO2@V2O5 cores can be tuned by controlling the number of ALD cycles. Catalytic methane oxidation experiments performed in a flow reactor at 600 ℃ demonstrate that SiO2@V2O5@Al2O3 nanostructures obtained after 50 ALD cycles exhibit the best catalytic activity (methane conversion = 22.2%; formaldehyde selectivity = 57.8%) and outperform all previously reported vanadium-based catalysts at 600 ℃. The prepared catalysts are subjected to in-depth characterization, which reveals that their Al2O3 shell provides new surfaces for the generation of highly disperse Td monomeric species with a V-O-Al bond by promoting interactions between Al2O3 and V2O5 nanoparticles during ALD. Moreover, the surface Al2O3 shell is found not only to protect V2O5 nanoparticles against sintering at 600 ℃, but also to anchor the produced Td monomeric vanadium species responsible for the high catalytic performance.
The stable tetrahedral geometry and high C-H bond dissociation energy of methane complicate its direct catalytic conversion; for example, the selective oxidation of methane to formaldehyde, which avoids the production of carbon dioxide by full oxidation and is therefore important for the versatile utilization of natural gas, is still viewed as challenging. Here, we utilize hydrothermal synthesis followed by atomic layer deposition (ALD) to prepare an efficient and thermally stable catalyst based on novel SiO2@V2O5@ Al2O3 core@shell nanostructures, showing that the thickness of Al2O3 shells over SiO2@V2O5 cores can be tuned by controlling the number of ALD cycles. Catalytic methane oxidation experiments performed in a flow reactor at 600 ℃ demonstrate that SiO2@V2O5@Al2O3 nanostructures obtained after 50 ALD cycles exhibit the best catalytic activity (methane conversion = 22.2%; formaldehyde selectivity = 57.8%) and outperform all previously reported vanadium-based catalysts at 600 ℃. The prepared catalysts are subjected to in-depth characterization, which reveals that their Al2O3 shell provides new surfaces for the generation of highly disperse Td monomeric species with a V-O-Al bond by promoting interactions between Al2O3 and V2O5 nanoparticles during ALD. Moreover, the surface Al2O3 shell is found not only to protect V2O5 nanoparticles against sintering at 600 ℃, but also to anchor the produced Td monomeric vanadium species responsible for the high catalytic performance.
62. Chemically impregnated NiO catalyst for molten electrolyte based gas-tank-free Li–O2 battery
Journal paper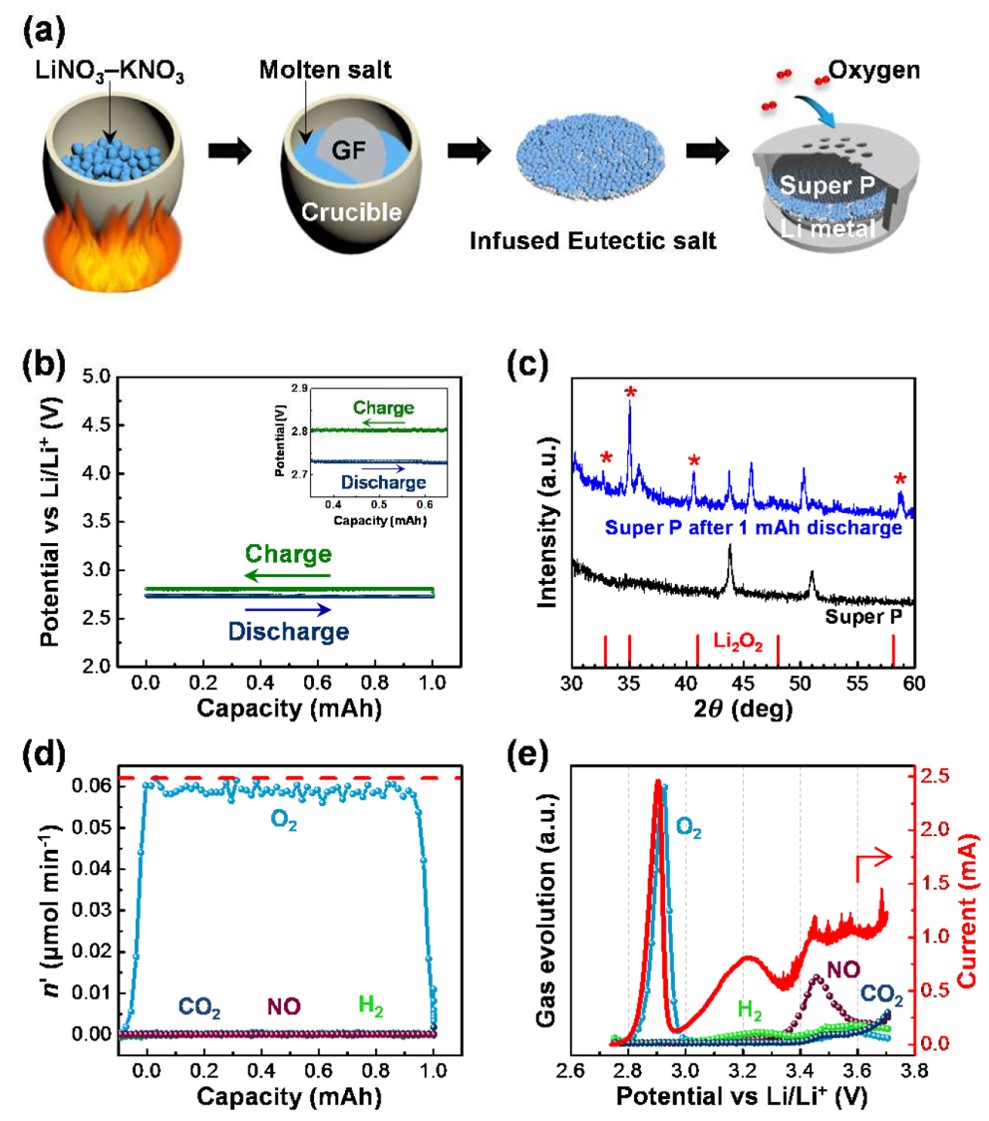
Closed Li-O2 batteries consisting of electrolytes derived from molten salt have emerged as attractive energy storage cells because of their unique oxygen-supply mechanism to form a stable Li2O discharge product without requiring an oxygen-gas-reservoir. However, the formation of stable Li2O discharge product increases the overpotential during the charging process, which compromises the cell performance because of the resulting parasitic reaction. In this study, we demonstrate a potent approach to reversibly operate an oxygen-gas-reservoir-free Li-O2 battery by using chemically impregnated nickel oxide (NiO) nanoparticles as a catalyst on the carbon electrode. The efficient bottom-up process for decorating NiO on a carbon material in binary molten electrolyte enables not only to significantly reduce the loading level of the catalyst but also to enhance the electrochemical performance with preventing the detrimental parasitic reaction in the oxygen-gas-reservoir-free Li-O2 cell. In particular, using the in situ gas analysis with electrochemical measurements, the 20 wt% NiO added to the carbon cathode is sufficient to reduce the charging potential without generation of parasitic gas evolution.
61. Design of Efficient Noble Metal Free Copper-promoted Nickel-Ceria-Zirconia Nanocatalyst for Bio-Fuel Upgrading
Journal paper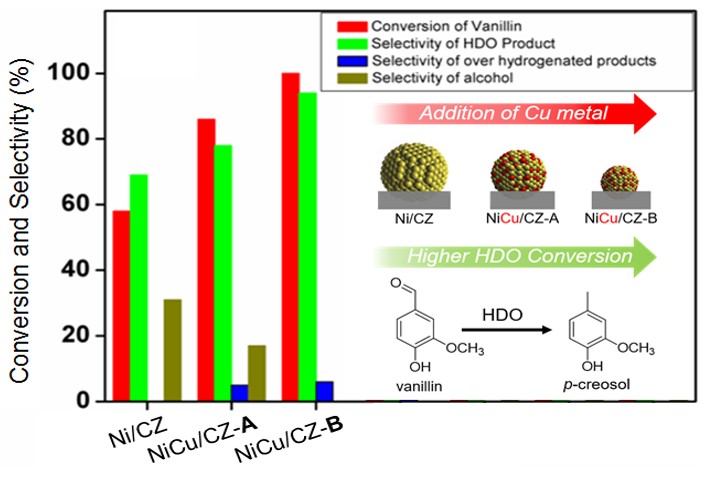 A new series of noble metal free bimetallic Cu-Ni supported ceria-zirconia catalysts (Cu-Ni/CZ) has been developed with various Cu/Ni ratios by a simple co-precipitation/impregnation method. Aqueous-phase hydrodeoxygenation (HDO) of vanillin a typical compound of lignin-derived bio-oil, in promoting biomass refining was carried out to investigate catalytic performances under 25 bar of H2 pressure at 160oC. All the as-synthesized catalysts were thoroughly characterised employing powder XRD, Raman spectroscopy, H2-TPR, HR-TEM, HAADF-STEM, EDS Mapping, and XPS. It was found that 15wt% Cu on Ni/CZ (Cu-Ni/CZ-B) nanocomposite enhanced activity significantly in comparison with other bimetallic or monometallic catalysts, exhibiting ~98% of vanillin conversion and ~94% of selectivity toward 2-methoxy-4-methylphenol as a desired product. The Cu-Ni/CZ-B catalyst shows ~4-fold increase in activity compared with Cu-Ni/CeO2 and Cu-Ni/ZrO2, the mono oxide supported counterparts. The superior catalytic performance with improved stability were explained by synergistic effects at the interfaces of each species, in which electron interactions between Ni, Cu, and ceria-zirconia support generated novel active sites. XRD and HR-TEM revealed that Cu-Ni bimetallic phases were created on the ceria-zirconia, which were responsible for enhancement of catalytic performance with no significant drop in catalytic activity with desired product selectivity for eleven successive catalytic cycles demonstrating the excellent stability and reproducibility of this catalyst system.
A new series of noble metal free bimetallic Cu-Ni supported ceria-zirconia catalysts (Cu-Ni/CZ) has been developed with various Cu/Ni ratios by a simple co-precipitation/impregnation method. Aqueous-phase hydrodeoxygenation (HDO) of vanillin a typical compound of lignin-derived bio-oil, in promoting biomass refining was carried out to investigate catalytic performances under 25 bar of H2 pressure at 160oC. All the as-synthesized catalysts were thoroughly characterised employing powder XRD, Raman spectroscopy, H2-TPR, HR-TEM, HAADF-STEM, EDS Mapping, and XPS. It was found that 15wt% Cu on Ni/CZ (Cu-Ni/CZ-B) nanocomposite enhanced activity significantly in comparison with other bimetallic or monometallic catalysts, exhibiting ~98% of vanillin conversion and ~94% of selectivity toward 2-methoxy-4-methylphenol as a desired product. The Cu-Ni/CZ-B catalyst shows ~4-fold increase in activity compared with Cu-Ni/CeO2 and Cu-Ni/ZrO2, the mono oxide supported counterparts. The superior catalytic performance with improved stability were explained by synergistic effects at the interfaces of each species, in which electron interactions between Ni, Cu, and ceria-zirconia support generated novel active sites. XRD and HR-TEM revealed that Cu-Ni bimetallic phases were created on the ceria-zirconia, which were responsible for enhancement of catalytic performance with no significant drop in catalytic activity with desired product selectivity for eleven successive catalytic cycles demonstrating the excellent stability and reproducibility of this catalyst system.
60. Boosting hot electron flux and catalytic activity at metal–oxide interfaces of PtCo bimetallic nanoparticles
Journal paper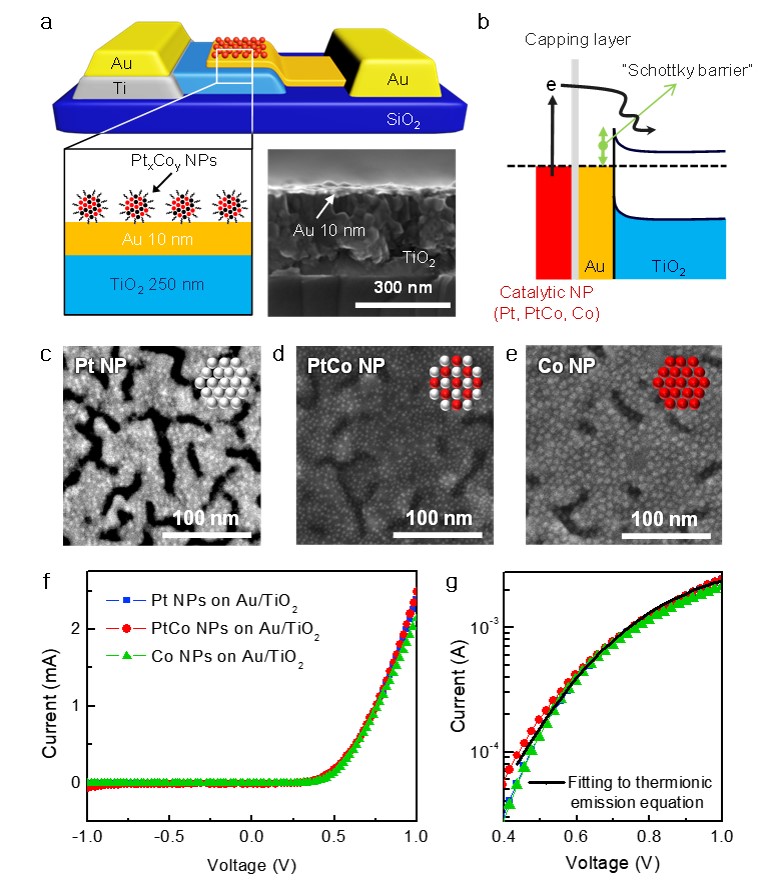 Despite numerous studies, the origin of the enhanced catalytic performance of bimetallic nanoparticles (NPs) remains elusive because of the ever-changing surface structures, compositions, and oxidation states of the NPs under reaction conditions. An effective strategy for attaining critical clues for the phenomenon is real-time quantitative detection of hot electrons induced by a chemical reaction on the catalysts. Here, we investigate hot electrons excited on PtCo bimetallic NPs during H2 oxidation by measuring the chemicurrent on a catalytic nanodiode while changing the Pt composition of the NPs. We reveal that the presence of a CoO/Pt interface enables efficient transport of electrons and higher catalytic activity for PtCo NPs. These results are consistent with theoretical calculations suggesting that lower activation energy and higher exothermicity are required for the reaction at the CoO/Pt interface.
Despite numerous studies, the origin of the enhanced catalytic performance of bimetallic nanoparticles (NPs) remains elusive because of the ever-changing surface structures, compositions, and oxidation states of the NPs under reaction conditions. An effective strategy for attaining critical clues for the phenomenon is real-time quantitative detection of hot electrons induced by a chemical reaction on the catalysts. Here, we investigate hot electrons excited on PtCo bimetallic NPs during H2 oxidation by measuring the chemicurrent on a catalytic nanodiode while changing the Pt composition of the NPs. We reveal that the presence of a CoO/Pt interface enables efficient transport of electrons and higher catalytic activity for PtCo NPs. These results are consistent with theoretical calculations suggesting that lower activation energy and higher exothermicity are required for the reaction at the CoO/Pt interface.
59. Acidic Effect of Porous Alumina as Supports for Pt Nanoparticle Catalysts in n-Hexane Reforming
Journal paper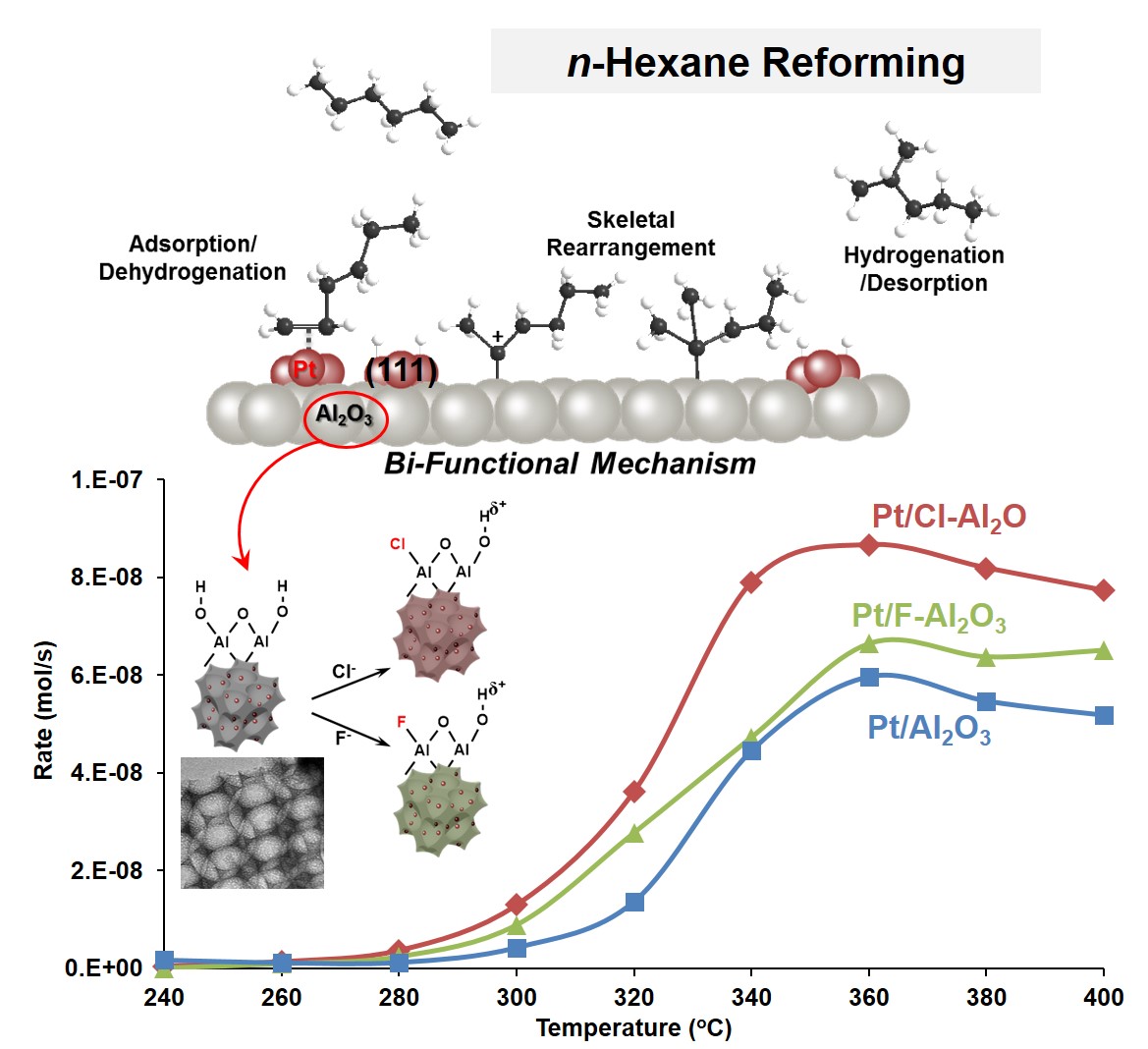 Acidic halogen-modified (Cl and F) porous alumina supports with well-defined macropores and mesopores were designed to prepare alumina-supported Pt nanoparticle (NP) catalysts (Pt/Cl-Al2O3, Pt/F-Al2O3, and Pt/Al2O3). The catalysts were then used for n-hexane reforming in a tubular fixed bed reactor with a hexane:H2 ratio of 1:4.3 at ambient pressure and various temperatures (240–400 °C). Although the reaction rates for all catalysts were maximised at 360 °C, Pt/Cl-Al2O3 exhibited the highest rate (at 8.66 × 10-8 mol/s). Regarding product selectivity, Pt/Cl-Al2O3 and Pt/F-Al2O3 yielded a higher number of olefin products and fewer cracking products than Pt/Al2O3. Temperature programmed desorption (TPD) with ethanol and in-situ diffuse-reflectance infrared spectroscopy (DRIFTS) with CO and pyridine adsorption were used to characterise the alumina support surface acidities. Compared with γ-Al2O3, the TPD results indicated that the Cl-Al2O3 and F-Al2O3 surface acidities were enhanced by surface modification. The in-situ DRIFTS experiments confirmed that the relative ratio of Lewis to Brønsted acid sites of Cl-Al2O3 (0.64) was higher than those of F-Al2O3 (0.54) and unmodified Al2O3 (0.56). Additionally, the DRIFTS spectra confirmed that the Pt NPs were preferentially deposited to the Lewis acid sites of the supports, and the CO adsorption spectra revealed that Pt NPs with (111) facets were preferentially deposited to the Lewis acid sites. The surface acidity studies indicated that the enhanced Lewis acidity of Cl-Al2O3 induced high reaction rates at all temperatures, resulting in skeletal rearrangements of hydrocarbons toward branched isomers at high temperature via a conventional bi-functional mechanism.
Acidic halogen-modified (Cl and F) porous alumina supports with well-defined macropores and mesopores were designed to prepare alumina-supported Pt nanoparticle (NP) catalysts (Pt/Cl-Al2O3, Pt/F-Al2O3, and Pt/Al2O3). The catalysts were then used for n-hexane reforming in a tubular fixed bed reactor with a hexane:H2 ratio of 1:4.3 at ambient pressure and various temperatures (240–400 °C). Although the reaction rates for all catalysts were maximised at 360 °C, Pt/Cl-Al2O3 exhibited the highest rate (at 8.66 × 10-8 mol/s). Regarding product selectivity, Pt/Cl-Al2O3 and Pt/F-Al2O3 yielded a higher number of olefin products and fewer cracking products than Pt/Al2O3. Temperature programmed desorption (TPD) with ethanol and in-situ diffuse-reflectance infrared spectroscopy (DRIFTS) with CO and pyridine adsorption were used to characterise the alumina support surface acidities. Compared with γ-Al2O3, the TPD results indicated that the Cl-Al2O3 and F-Al2O3 surface acidities were enhanced by surface modification. The in-situ DRIFTS experiments confirmed that the relative ratio of Lewis to Brønsted acid sites of Cl-Al2O3 (0.64) was higher than those of F-Al2O3 (0.54) and unmodified Al2O3 (0.56). Additionally, the DRIFTS spectra confirmed that the Pt NPs were preferentially deposited to the Lewis acid sites of the supports, and the CO adsorption spectra revealed that Pt NPs with (111) facets were preferentially deposited to the Lewis acid sites. The surface acidity studies indicated that the enhanced Lewis acidity of Cl-Al2O3 induced high reaction rates at all temperatures, resulting in skeletal rearrangements of hydrocarbons toward branched isomers at high temperature via a conventional bi-functional mechanism.
58. Specific Metal–Support Interactions between Nanoparticle Layers for Catalysts with Enhanced Methanol Oxidation Activity
Journal paper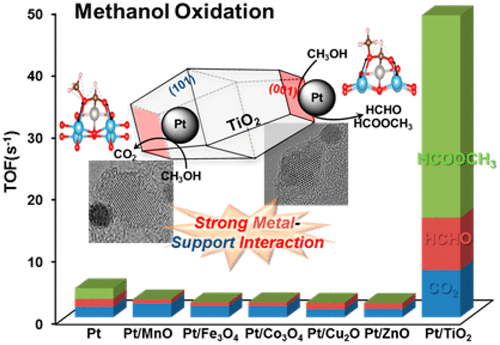 Oxide supports often play a critical role in metal-supported catalysts owing to their charge transfer phenomena that can alter catalytic performance. Herein, in place of conventional bulk oxide supports, monodisperse oxide nanoparticles (NPs) were exploited as supports for Pt catalysts. Depending on the type of oxide NP, Pt/oxide layered catalysts exhibited dramatic changes in the catalytic activity and selectivity for methanol oxidation. While Pt NPs deposited on MnO, Fe3O4, Co3O4, Cu2O, and ZnO NPs had comparable turnover frequencies to that of the pure Pt NP catalyst, Pt deposited TiO2 NPs changed the reaction rate significantly, with preferential selectivity observed toward partial oxidation products. Facet-specific interactions between Pt and TiO2 NPs were demonstrated by density functional theory calculations and catalytic reactions using shape-controlled TiO2 NPs. When Pt NPs were attached to spherical and rhombic TiO2 NPs with abundant (001) surfaces, methanol conversion was enhanced 10-fold owing to strong charge transfer from TiO2 to Pt.
Oxide supports often play a critical role in metal-supported catalysts owing to their charge transfer phenomena that can alter catalytic performance. Herein, in place of conventional bulk oxide supports, monodisperse oxide nanoparticles (NPs) were exploited as supports for Pt catalysts. Depending on the type of oxide NP, Pt/oxide layered catalysts exhibited dramatic changes in the catalytic activity and selectivity for methanol oxidation. While Pt NPs deposited on MnO, Fe3O4, Co3O4, Cu2O, and ZnO NPs had comparable turnover frequencies to that of the pure Pt NP catalyst, Pt deposited TiO2 NPs changed the reaction rate significantly, with preferential selectivity observed toward partial oxidation products. Facet-specific interactions between Pt and TiO2 NPs were demonstrated by density functional theory calculations and catalytic reactions using shape-controlled TiO2 NPs. When Pt NPs were attached to spherical and rhombic TiO2 NPs with abundant (001) surfaces, methanol conversion was enhanced 10-fold owing to strong charge transfer from TiO2 to Pt.
57. Supported Pd nanoparticle catalysts with high activities and selectivities in liquid-phase furfural hydrogenation
Journal paper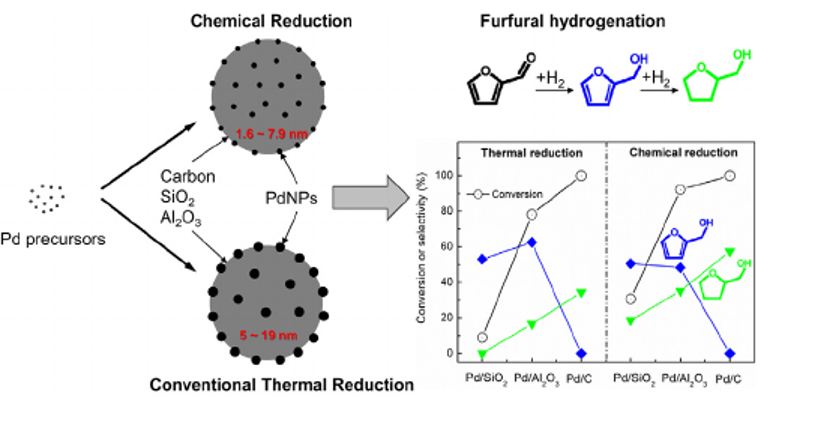 Highly dispersed and ultrafine Pd nanoparticles (NPs) deposited on carbon, silica, and alumina supports were prepared by chemical reduction (CR) using sodium borohydride, in the presence of trisodium citrate as a stabilizer. For comparison, supported Pd catalysts were also prepared through impregnation followed by thermal reduction (TR) and capillary inclusion of the colloidal Pd NPs (CI). The activities and selectivities of the prepared catalysts were evaluated in the liquid-phase furfural (FAL) hydrogenation reaction under 20 bar H2 at 180 °C. Under these conditions, FAL was converted via two distinct pathways to produce either furan via decarbonylation or furfuryl alcohol (FA) via aldehyde hydrogenation. Subsequently, furan and FA were converted to tetrahydrofuran and tetrahydrofurfuryl alcohol (THFA), respectively, via ring hydrogenation. 2-Methylfuran was also produced from the hydration of FAL. To verify the efficiency of the preparation methods, the size of the Pd NPs, the degree of metal dispersion, and the type of supports were correlated with the catalytic conversions and selectivities of FAL hydrogenation. It was confirmed that the 5 wt% Pd/C catalysts possessed highly dispersed small Pd NPs with large metallic Pd surface areas, which resulted in high conversions and selectivities towards THFA in the FAL hydrogenation reaction compared to conventional supported catalysts.
Highly dispersed and ultrafine Pd nanoparticles (NPs) deposited on carbon, silica, and alumina supports were prepared by chemical reduction (CR) using sodium borohydride, in the presence of trisodium citrate as a stabilizer. For comparison, supported Pd catalysts were also prepared through impregnation followed by thermal reduction (TR) and capillary inclusion of the colloidal Pd NPs (CI). The activities and selectivities of the prepared catalysts were evaluated in the liquid-phase furfural (FAL) hydrogenation reaction under 20 bar H2 at 180 °C. Under these conditions, FAL was converted via two distinct pathways to produce either furan via decarbonylation or furfuryl alcohol (FA) via aldehyde hydrogenation. Subsequently, furan and FA were converted to tetrahydrofuran and tetrahydrofurfuryl alcohol (THFA), respectively, via ring hydrogenation. 2-Methylfuran was also produced from the hydration of FAL. To verify the efficiency of the preparation methods, the size of the Pd NPs, the degree of metal dispersion, and the type of supports were correlated with the catalytic conversions and selectivities of FAL hydrogenation. It was confirmed that the 5 wt% Pd/C catalysts possessed highly dispersed small Pd NPs with large metallic Pd surface areas, which resulted in high conversions and selectivities towards THFA in the FAL hydrogenation reaction compared to conventional supported catalysts.
56. Catalytic 1-Propanol Oxidation on Size-Controlled Platinum Nanoparticles at Solid–Gas and Solid–Liquid Interfaces: Significant Differences in Kinetics and Mechanisms
Journal paper Utilizing Pt nanoparticles of varying sizes (2–7 nm), it was found that the oxidation of 1-propanol by molecular oxygen at 60 °C to propanal at the solid–gas and solid–liquid interfaces yielded significantly different results depending on Pt particle size and alcohol surface density. The reaction rate at the solid–gas interface was found to be 1 order of magnitude greater than that at the solid–liquid interface after normalizing concentration. In addition, catalytic activity increases with the size of Pt nanoparticles for both reactions. Moreover, water substantially promoted 1-propanol oxidation in the liquid phase, yet it inhibited the reaction in the gas phase. The gas phase and liquid phase reactions are believed to undergo different mechanisms due to differing kinetic results. This correlated well with different orientations of the 1-propanol species at the solid–gas interface versus the solid–liquid interface as probed by sum–frequency generation vibrational spectroscopy (SFGVS) under reaction conditions and simulated by computational density function theory calculations.
Utilizing Pt nanoparticles of varying sizes (2–7 nm), it was found that the oxidation of 1-propanol by molecular oxygen at 60 °C to propanal at the solid–gas and solid–liquid interfaces yielded significantly different results depending on Pt particle size and alcohol surface density. The reaction rate at the solid–gas interface was found to be 1 order of magnitude greater than that at the solid–liquid interface after normalizing concentration. In addition, catalytic activity increases with the size of Pt nanoparticles for both reactions. Moreover, water substantially promoted 1-propanol oxidation in the liquid phase, yet it inhibited the reaction in the gas phase. The gas phase and liquid phase reactions are believed to undergo different mechanisms due to differing kinetic results. This correlated well with different orientations of the 1-propanol species at the solid–gas interface versus the solid–liquid interface as probed by sum–frequency generation vibrational spectroscopy (SFGVS) under reaction conditions and simulated by computational density function theory calculations.
 CO oxidation, as one of the simplest catalytic reactions, has been widely studied in heterogeneous catalysis. Since CO can be easily adsorbed on active metals like Au, Pt, Pd, Rh, and Ru, many researchers have attempted to observe surface phenomena, including adsorption, surface restructuring, and oxidation kinetics on diverse metal surfaces ranging from single crystals to nanoparticles (NPs). The rapid advances in nanochemistry have further stimulated studies of catalytic reactions, and in-depth understanding of CO oxidation is possible based on analyses of how the size, shape, and composition affect the activity and determine the most effective active site of metal NPs. Recent research has demonstrated that the CO oxidation activity varies with the size of metallic NPs. The oxidation state of the NPs varies as a function of the size, and the surface oxide layers formed on NPs have been found to be important in enhancing or suppressing CO oxidation. Surface segregation of bimetallic NPs also influences the CO oxidation rate. The NP surfaces undergo significant adsorbate-induced structural changes, as confirmed by various in situ characterization techniques under catalytically relevant CO oxidation conditions. Based on the knowledge of support-induced catalytic properties, various metal-support interactions have been investigated for enhancing CO oxidation. By changing the reaction environments to either CO- or O-rich atmospheres, the synergetic effect at the interfaces of NPs and support oxides have been largely clarified. Versatile nanostructures with confined shells of small metal NPs have also been designed as core@shell-type catalysts. Using in situ characterization techniques combined with well-defined NPs, it is possible to study CO oxidation on the molecular level in order to gain vital mechanistic insights under catalytic working conditions.
CO oxidation, as one of the simplest catalytic reactions, has been widely studied in heterogeneous catalysis. Since CO can be easily adsorbed on active metals like Au, Pt, Pd, Rh, and Ru, many researchers have attempted to observe surface phenomena, including adsorption, surface restructuring, and oxidation kinetics on diverse metal surfaces ranging from single crystals to nanoparticles (NPs). The rapid advances in nanochemistry have further stimulated studies of catalytic reactions, and in-depth understanding of CO oxidation is possible based on analyses of how the size, shape, and composition affect the activity and determine the most effective active site of metal NPs. Recent research has demonstrated that the CO oxidation activity varies with the size of metallic NPs. The oxidation state of the NPs varies as a function of the size, and the surface oxide layers formed on NPs have been found to be important in enhancing or suppressing CO oxidation. Surface segregation of bimetallic NPs also influences the CO oxidation rate. The NP surfaces undergo significant adsorbate-induced structural changes, as confirmed by various in situ characterization techniques under catalytically relevant CO oxidation conditions. Based on the knowledge of support-induced catalytic properties, various metal-support interactions have been investigated for enhancing CO oxidation. By changing the reaction environments to either CO- or O-rich atmospheres, the synergetic effect at the interfaces of NPs and support oxides have been largely clarified. Versatile nanostructures with confined shells of small metal NPs have also been designed as core@shell-type catalysts. Using in situ characterization techniques combined with well-defined NPs, it is possible to study CO oxidation on the molecular level in order to gain vital mechanistic insights under catalytic working conditions.
54. Transition Metal-Based Thiometallates as Surface Ligands for Functionalization of All-Inorganic Nanocrystals
Journal paper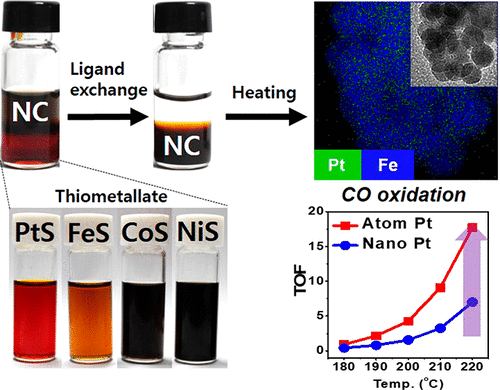 We report a new family of inorganic ligands, namely, transition metal-based thiometallates, for the surface functionalization of colloidal nanocrystals (NCs). We synthesized Pt-, Fe-, Co-, and Ni-based thiometallates, in which transition metal ions were complexed with polysulfides. These inorganic anions easily exchanged the surface organic ligands of various nanocrystals of metal, semiconductor, and oxide materials, without affecting the NCs’ primary structural and optical characteristics. Furthermore, upon heating, these complexes were decomposed and transformed into crystalline phases of metal sulfides or pure metals, accompanied by the evaporation of S. Based on this effect, we selectively synthesized homogeneously distributed atomic Pt clusters or Pt nanoparticles on Fe3O4 nanomaterials by heating thioplatinate-capped Fe3O4 NCs. As a model application, we tested the prepared Pt-functionalized Fe3O4 nanomaterial as a heterogeneous catalyst for CO oxidation reaction and Pt–Fe3O4 catalysts exhibited the high turnover frequency due to the homogeneous distribution of atomic Pt over Fe3O4 and the corresponding strong metal–support interaction. This approach opens up a new avenue to functionalize nanocrystals for catalytic applications.
We report a new family of inorganic ligands, namely, transition metal-based thiometallates, for the surface functionalization of colloidal nanocrystals (NCs). We synthesized Pt-, Fe-, Co-, and Ni-based thiometallates, in which transition metal ions were complexed with polysulfides. These inorganic anions easily exchanged the surface organic ligands of various nanocrystals of metal, semiconductor, and oxide materials, without affecting the NCs’ primary structural and optical characteristics. Furthermore, upon heating, these complexes were decomposed and transformed into crystalline phases of metal sulfides or pure metals, accompanied by the evaporation of S. Based on this effect, we selectively synthesized homogeneously distributed atomic Pt clusters or Pt nanoparticles on Fe3O4 nanomaterials by heating thioplatinate-capped Fe3O4 NCs. As a model application, we tested the prepared Pt-functionalized Fe3O4 nanomaterial as a heterogeneous catalyst for CO oxidation reaction and Pt–Fe3O4 catalysts exhibited the high turnover frequency due to the homogeneous distribution of atomic Pt over Fe3O4 and the corresponding strong metal–support interaction. This approach opens up a new avenue to functionalize nanocrystals for catalytic applications.
53. Postsynthesis Modulation of the Catalytic Interface inside a Hollow Nanoreactor: Exploitation of the Bidirectional Behavior of Mixed-Valent Mn3O4 Phase in the Galvanic Replacement Reaction.
Journal paper49
This paper proposes a postsynthesis strategy to modulate the synergy of the catalyst/support interface of the preformed hollow nanoreactor by replacing preloaded support material inside the cavity, which therefore enhances the variability of the nanoreactor approach. This strategy exploits the newly explored bidirectional behavior of an Mn3O4 nanoparticle of mixed-valent state, which either sequentially or simultaneously templates reduction of noble-metal ions and oxidation of Ce3+ ions during galvanic replacement. The application of this process to the modification of the preformed hollow silica nanoreactor led to the replacement of the catalyst-immobilizing Mn3O4 layer inside the cavity with CeO2 while preserving the tiny size and well-dispersed state of supported catalysts and, as a result, allowing for the introduction of a synergistic noble-metal/CeO2 interface exerting the highly improved catalytic performance in CO oxidation reaction.
52. Photocatalytic H2 generation on macro-mesoporous oxide-supported Pt nanoparticles
Journal paperA series of p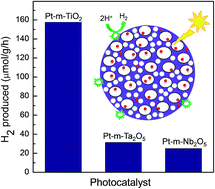
51. Soft X-ray spectroscopy studies of adsorption and reaction of CO in the presence of H2 over 6 nm MnO nanoparticles supported on mesoporous Co3O4
Journal paper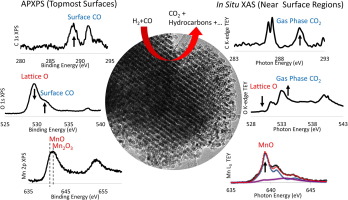 MnO nanoparticles (6 nm) were supported on mesoporous spinel Co3O4 and studied using ambient pressure X-ray photoelectron spectroscopy (APXPS) and in situ X-ray absorption spectroscopy (XAS) during hydrogenation of CO. The nature and evolution of surface adsorbed species as well as the oxidation states of the metal oxide surfaces were evaluated under oxidizing, reducing, and H2 + CO (2:1) reaction atmospheres. From APXPS, MnO nanoparticle surfaces were found to be progressively reduced in H2 atmospheres with increasing temperature.
MnO nanoparticles (6 nm) were supported on mesoporous spinel Co3O4 and studied using ambient pressure X-ray photoelectron spectroscopy (APXPS) and in situ X-ray absorption spectroscopy (XAS) during hydrogenation of CO. The nature and evolution of surface adsorbed species as well as the oxidation states of the metal oxide surfaces were evaluated under oxidizing, reducing, and H2 + CO (2:1) reaction atmospheres. From APXPS, MnO nanoparticle surfaces were found to be progressively reduced in H2 atmospheres with increasing temperature.
Since 2015 at UNIST
50. High-performance hybrid oxide catalyst of manganese and cobalt for low-pressure methanol synthesis
Journal paper
Carbon dioxide capture and use as a carbon feedstock presents both environmental and industrial benefits. Here we report the discovery of a hybrid oxide catalyst comprising manganese oxide nanoparticles supported on mesoporous spinel cobalt oxide, which catalyses the conversion of carbon dioxide to methanol at high yields. In addition, carbon–carbon bond formation is observed through the production of ethylene. We document the existence of an active interface between cobalt oxide surface layers and manganese oxide nanoparticles by using X-ray absorption spectroscopy and electron energy-loss spectroscopy in the scanning transmission electron microscopy mode. Through control experiments, we find that the catalyst’s chemical nature and architecture are the key factors in enabling the enhanced methanol synthesis and ethylene production. To demonstrate the industrial applicability, the catalyst is also run under high conversion regimes, showing its potential as a substitute for current methanol synthesis technologies.
49. Nanocatalysis I: Synthesis of metal and bimetallic nanoparticles and porous oxides and their catalytic reaction studies
Journal paperIn recent heterogeneous catalysis, much effort has been made in understanding how the size, shape, and composition of nanoparticles and oxide-metal interfaces affect catalytic performance at the molecular level. Recent advances in colloidal synthetic techniques enable preparing diverse metallic or bimetallic nanoparticles with welldefined size, shape, and composition and porous oxides as
a high surface support. As nanoparticles become smaller, new chemical, physical, and catalytic properties emerge.
Geometrically, as the smaller the nanoparticle the greater the relative number of edge and corner sites per unit surface of the nanoparticle. When the nanoparticles are smaller than a critical size (2.7 nm), finite-size effects such as a change of adsorption strength or oxidation state are revealed by changes in their electronic structures. By alloying two metals, the formation of heteroatom bonds and geometric effects such as strain due to the change of metal–metal bond lengths cause new electronic structures to appear in bimetallic nanoparticles. Ceaseless catalytic reaction studies have been discovered that the highest reaction yields, product selectivity, and process stability were achieved by determining the critical size, shape, and composition of nanoparticles and by choosing the appropriate oxide support. Depending on the pore size, various kinds of micro-, meso-, and macro-porous materials are fabricated by the aid of structure-directing agents or hardtemplates.
Recent achievements for the preparation of versatile core/shell nanostructures composing mesoporous oxides, zeolites, and metal organic frameworks provide new insights toward nanocatalysis with novel ideas.
48. Hollow MnOxPy and Pt/MnOxPy yolk/shell nanoparticles as a T1 MRI contrast agent
Journal paper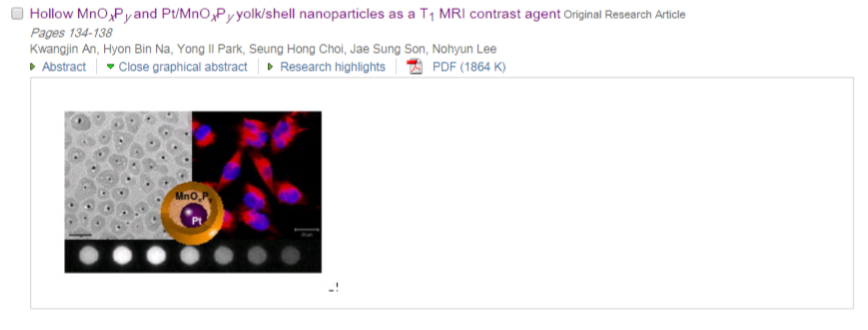
Hollow MnOxPy and Pt/MnOxPy yolk/shell nanoparticles were fabricated via the nanoscale acid-etching process from the solid MnO and Pt/MnO core/shell nanoparticles, respectively. In the synthesis, alkylphosphonic acid impurity in trioctylphosphine oxide was a key component, resulting in amorphous hollow metal phosphate nanoparticles. Hollow nanoparticles containing Mn2+ ions showed positively enhanced T1 relaxation, in which the r1 values of the hollow MnOxPy and Pt/MnOxPy yolk/shell were greater than that of the original MnO nanocrystals. The increased surface area of the hollow nanoparticles enhanced interaction between Mn2+ ions of the nanoparticle surface and water molecules. Cytotoxicity experiment revealed that Mn ions released from hollow walls of the MnOxPy nanoparticles were responsible for the cytotoxicity, while Pt ions from the Pt/MnOxPy yolk/shell were not released in the cells. These nanoparticles provide potential insights into an anticancer drug, enabling simultaneous T1 magnetic resonance imaging (MRI) and therapy.
47. Comparing the Catalytic Oxidation of Ethanol at the Solid−Gas and Solid−Liquid Interfaces over Size-Controlled Pt Nanoparticles: Striking Differences in Kinetics and Mechanism
Journal paper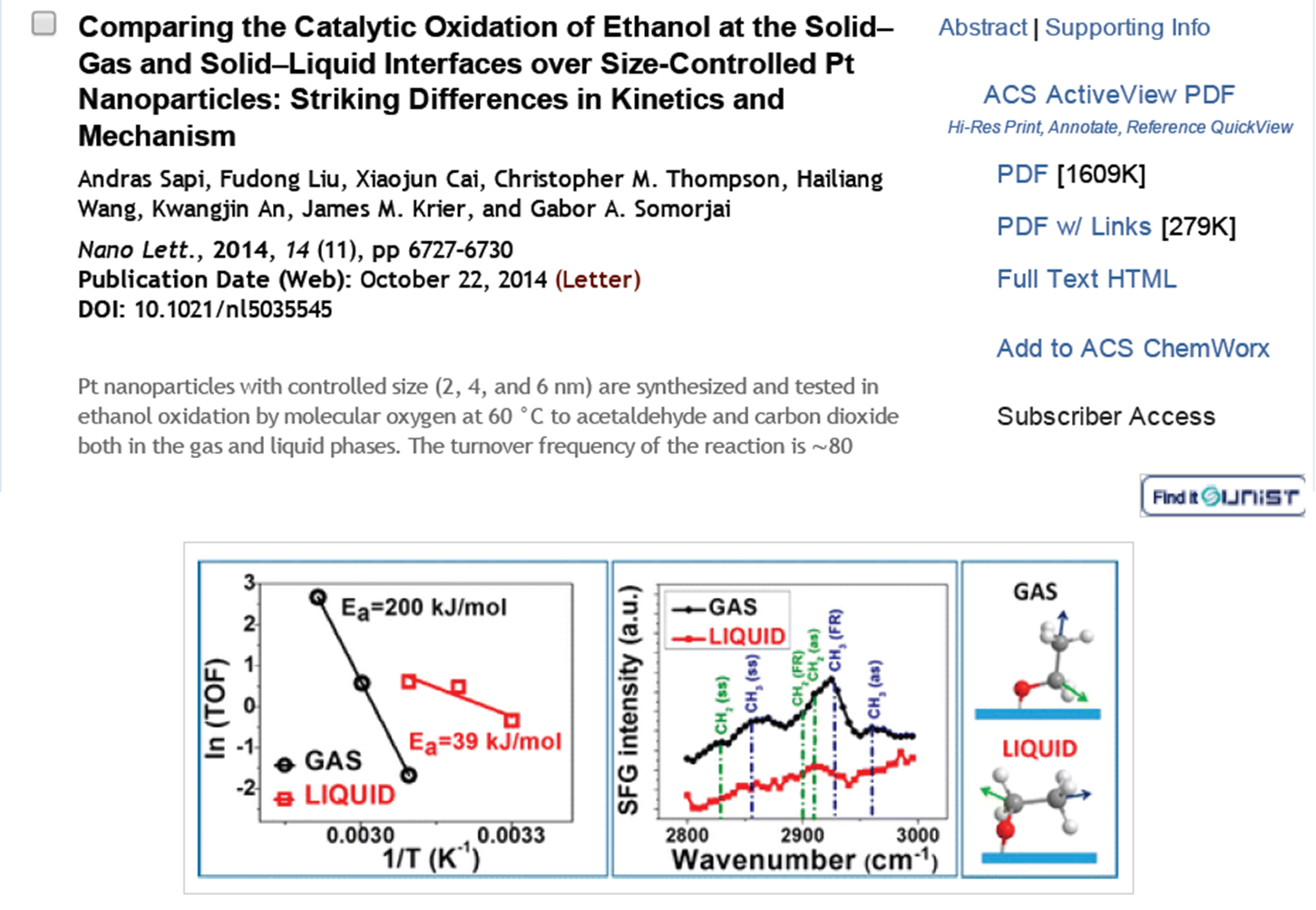
Pt nanoparticles with controlled size (2, 4, and 6 nm) are synthesized and tested in ethanol oxidation by molecular oxygen at 60 °C to acetaldehyde and carbon dioxide both in the gas and liquid phases. The turnover frequency of the reaction is ∼80 times faster, and the activation energy is ∼5 times higher at the gas–solid interface compared to the liquid–solid interface. The catalytic activity is highly dependent on the size of the Pt nanoparticles; however, the selectivity is not size sensitive. Acetaldehyde is the main product in both media, while twice as much carbon dioxide was observed in the gas phase compared to the liquid phase. Added water boosts the reaction in the liquid phase; however, it acts as an inhibitor in the gas phase. The more water vapor was added, the more carbon dioxide was formed in the gas phase, while the selectivity was not affected by the concentration of the water in the liquid phase. The differences in the reaction kinetics of the solid–gas and solid–liquid interfaces can be attributed to the molecular orientation deviation of the ethanol molecules on the Pt surface in the gas and liquid phases as evidenced by sum frequency generation vibrational spectroscopy.
46. Effects of Nanoparticle Size and Metal/Support Interactions in Pt-Catalyzed Methanol Oxidation Reactions in Gas and Liquid Phases
Journal paperWe compare catalytic methanol oxidation reactions in the gas and liquid phases by focusing on the kinetic effects of platinum nanoparticle size and metal/support interactions. Under the reaction conditions at 60 °C, methanol can be oxidized to multiple products including carbon dioxide (full-oxidation product), formaldehyde (partial-oxidation product) and methyl formate (partial-oxidation and coupling product). We use 2, 4, 6 and 8 nm platinum nanoparticles supported on mesoporous silica as catalysts to study the size effect, and 2.5 nm platinum nanoparticles supported on mesoporousSiO2, Co3O4, MnO2, Fe2O3, NiO and CeO2 to study the metal/oxide interface effect. We find that all three products are formed with comparable selectivities in the gas phase, but in the liquid phase formaldehyde is the dominant product. While the influence of size on activity is not substantial in the gas phase, the liquid-phase reaction rates monotonically increase by a factor of 6 in the size range of 2–8 nm. The reaction rates in the gas phase are dramatically affected by the strong interactions between the platinum nanoparticles and transition metal oxide supports. While the Pt/MnO2 is 135 times less active, the Pt/CeO2 is 12 times more active, both compared to the Pt/SiO2. However in the liquid phase, the support effect is less significant, with the most active catalyst Pt/MnO2 exhibiting an enhancement factor of 2.5 compared to the Pt/SiO2. Our results suggest that the kinetic effects of platinum nanoparticle size and metal/support interactions can be totally different between the solid/gas and solid/liquid interfaces even for the same chemical reaction.
45. High-Temperature Catalytic Reforming of n‑Hexane over Supported and Core−Shell Pt Nanoparticle Catalysts: Role of Oxide−Metal Interface and Thermal Stability
Journal paper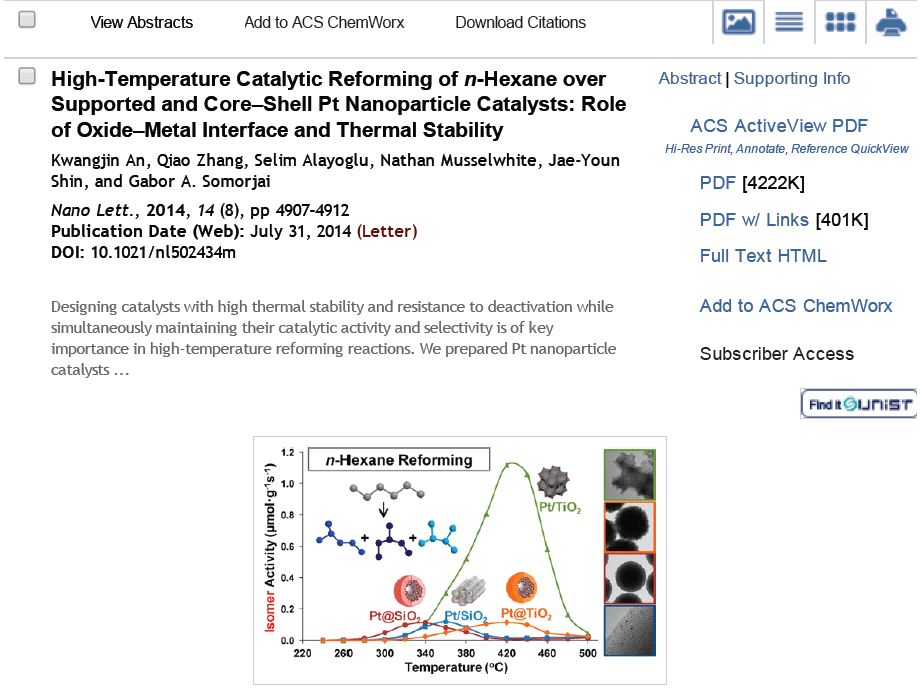
Selective Designing catalysts with high thermal stability and resistance to deactivation while simultaneously maintaining their catalytic activity and selectivity is of key importance in hightemperature reforming reactions. We prepared Pt nanoparticle catalysts supported on either mesoporous SiO2 or TiO2. Sandwich-type Pt core@shell catalysts (SiO2@Pt@SiO2 and SiO2@Pt@TiO2) were also synthesized from Pt nanoparticles deposited on SiO2 spheres, which were encapsulated by either mesoporous SiO2 or TiO2 shells. n-Hexane reforming was carried out over these four catalysts at 240−500 °C with a hexane/H2 ratio of 1:5 to investigate thermal stability and the role of the support. For the production of high-octane gasoline, branched C6 isomers are more highly desired than other cyclic, aromatic, and
cracking products. Over Pt/TiO2 catalyst, production of 2-methylpentane and 3-methylpentane via isomerization was increased selectively up to 420 °C by charge transfer at Pt−TiO2 interfaces, as compared to Pt/SiO2. When thermal stability was compared between supported catalysts and sandwich-type core@shell catalysts, the Pt/SiO2 catalyst suffered sintering above 400 °C, whereas the SiO2@Pt@SiO2 catalyst preserved the Pt nanoparticle size and shape up to 500 °C. The SiO2@Pt@TiO2 catalyst led to Pt nanoparticle sintering due to incomplete protection of the TiO2 shells during the reaction at 500 °C. Interestingly, over thePt/TiO2 catalyst, the average size of Pt nanoparticles was maintained even after 500 °C without sintering. In situ ambient pressure X-ray photoelectron spectroscopy demonstrated that the Pt/TiO2 catalyst did not exhibit TiO2 overgrowth on the Pt surface or deactivation by Pt sintering up to 600 °C. The extraordinarily high stability of the Pt/TiO2 catalyst promoted high reaction rates, which was 8 times greater than other catalysts and high isomer selectivity. By the strong metal−support interaction, the Pt/TiO2 was turned out as the best catalyst with great thermal stability as well as high reaction rate and product selectivity in high-temperature reforming reaction.
44. CO oxidation on PtSn nanoparticle catalysts occurs at the interface of Pt and Sn oxide domains formed under reaction conditions
Journal paper
The barrier to CO oxidation on Pt catalysts is the strongly bound adsorbed CO, which inhibits O2 adsorption and hinders CO2 formation. Using reaction studies and in situ X-ray spectroscopy with colloidally prepared, monodisperse ~2 nm Pt and PtSn nanoparticle catalysts, we show that the addition of Sn to Pt provides distinctly different reaction sites and a more efficient reaction mechanism for CO oxidation compared to pure Pt catalysts. To probe the influence of Sn, we intentionally poisoned the Pt component of the nanoparticle catalysts using a CO-rich atmosphere. With a reaction environment comprised of 100 Torr CO and 40 Torr O2 and a temperature range between 200 and 300 C, Pt and PtSn catalysts exhibited activation barriers for CO2 formation of 133 kJ/mol and 35 kJ/mol, respectively. While pure Sn is readily oxidized and is not active for CO oxidation, the addition of Sn to Pt provides an active site for O2 adsorption that is important when Pt is covered with CO. Sn oxide was identified as the active Sn species under reaction conditions by in situ ambient pressure X-ray photoelectron spectroscopy measurements. While chemical signatures of Pt and Sn indicated intermixed metallic components under reducing conditions, Pt and Sn were found to reversibly separate into isolated domains of Pt and oxidic Sn on the nanoparticle surface under reaction conditions of 100 mTorr CO and 40 mTorr O2 between temperatures of 200-275 C. Under these conditions, PtSn catalysts exhibited apparent reaction orders in O2 for CO2 production that were 0.5 and lower with increasing partial pressures. These reaction orders contrast the first-order dependence in O2 known for pure Pt. The differences in activation barriers, non-first-order dependence in O2, and the presence of a partially oxidized Sn indicate that the enhanced activity is due to a reaction mechanism that occurs at a Pt/Sn oxide interface present at the nanoparticle surface.
43. Designed Catalysts from Pt Nanoparticles Supported on Macroporous Oxides for Selective Isomerization of n-Hexane
Journal paper
Selective isomerization toward branched hydrocarbons is an important catalytic process in oil refining to obtain high-octane gasoline with minimal content of aromatic compounds. Colloidal Pt nanoparticles with controlled sizes of 1.7, 2.7, and 5.5 nm were deposited onto ordered macroporous oxides of SiO2, Al2O3, TiO2, Nb2O5, Ta2O5, and ZrO2 to investigate Pt size- and support-dependent catalytic selectivity in n-hexane isomerization. Among the macroporous oxides, Nb2O5 and Ta2O5 exhibited the highest product selectivity, yielding predominantly branched C6 isomers, including 2- or 3-methylpentane, as desired products ofn-hexane isomerization (140 Torr n-hexane and 620 Torr H2 at 360 °C). In situ characterizations including X-ray diffraction and ambient-pressure X-ray photoelectron spectroscopy showed that the crystal structures of the oxides in Pt/oxide catalysts were not changed during the reaction and oxidation states of Nb2O5 were maintained under both H2and O2 conditions. Fourier transform infrared spectra of pyridine adsorbed on the oxides showed that Lewis sites were the dominant acidic site of the oxides. Macroporous Nb2O5 and Ta2O5 were identified to play key roles in the selective isomerization by charge transfer at Pt–oxide interfaces. The selectivity was revealed to be Pt size-dependent, with improved isomer production as Pt sizes increased from 1.7 to 5.5 nm. When 5.5 nm Pt nanoparticles were supported on Nb2O5 or Ta2O5, the selectivity toward branched C6 isomers was further increased, reaching ca. 97% with a minimum content of benzene, due to the combined effects of the Pt size and the strong metal–support interaction.
42. Evidence of Highly Active Cobalt Oxide Catalyst for the Fischer−Tropsch Synthesis and CO2 Hydrogenation
Journal paper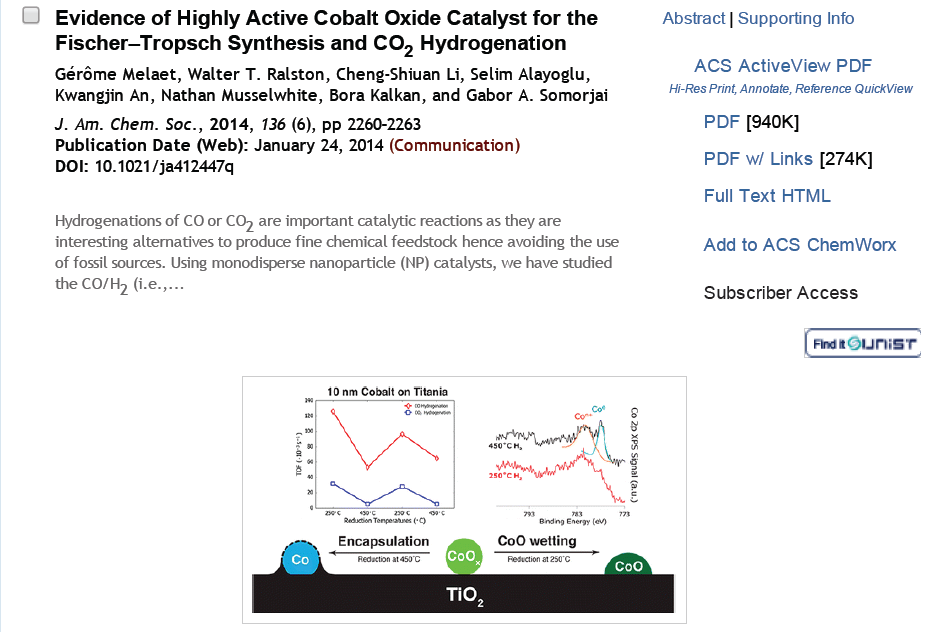
Hydrogenations of CO or CO2 are important catalytic reactions as they are interesting alternatives to produce fine chemical feedstock hence avoiding the use of fossil sources. Using monodisperse nanoparticle (NP) catalysts, we have studied the CO/H2 (i.e., Fischer-Tropsch synthesis) and CO2/H2 reactions. Exploiting synchrotron based in situ characterization techniques such as XANES and XPS, we were able to demonstrate that 10 nm Co NPs cannot be reduced at 250 °C while supported on TiO2 or SiO2 and that the complete reduction of cobalt can only be achieved at 450 °C. Interestingly, cobalt oxide performs better than fully reduced cobalt when supported on TiO2. In fact, the catalytic results indicate an enhancement of 10-fold for the CO2/H2 reaction rate and 2-fold for the CO/H2 reaction rate for the Co/TiO2 treated at 250 °C in H2 versus Co/TiO2 treated at 450 °C. Inversely, the activity of cobalt supported on SiO2 has a higher turnover frequency when cobalt is metallic. The product distributions could be tuned depending on the support and the oxidation state of cobalt. For oxidized cobalt on TiO2, we observed an increase of methane production for the CO2/H2 reaction whereas it is more selective to unsaturated products for the CO/H2 reaction. In situ investigation of the catalysts indicated wetting of the TiO2 support by CoOx and partial encapsulation of metallic Co by TiO2-x.
41. Spin excitations in cubic maghemite nanoparticles studied by time-of-flight neutron spectroscopy.
Journal paperWe have determined the field dependence of collective magnetic excitations in iron oxide nanoparticles of cubic shape with 8.42(2) nm edge length and a narrow log normal size distribution of 8.2(2)% using time-of-flight neutron spectroscopy. The energy dependence of the uniform precession modes was investigated up to 5 T applied field and yields a Land´e factor g = 2.05(2) as expected for maghemite (γ -Fe2O3) nanoparticles. A large effective anisotropy field of BA,eff = 0.45(16) T was determined, in excellent agreement with macroscopic measurements. This anisotropy is attributed to enhanced shape anisotropy in these monodisperse cubic nanoparticles. The combination of our results with macroscopic magnetization information provides a consistent view of the energyscales of superparamagnetic relaxation and collective magnetic excitations in magnetic nanoparticles.
40. Pt-mediated Reversible Reduction and Expansion of CeO2 in Pt Nanoparticle/mesoporous CeO2 Catalyst: In situ X-ray Spectroscopy and Diffraction Studies under Redox (H2 and O2) Atmospheres.
Journal paper
Here, we report the Pt nanoparticle mediated reduction (oxidation) and lattice expansion (contraction) of mesoporous CeO2 under H2 (O2) atmospheres and in the temperature range of 50−350 °C. We found that CeO2 in the Pt/CeO2 catalyst was partially reduced in H2 (and fully oxidized back in O2) as demonstrated by several in situ techniques: APXPS spectra (4d core levels) for the topmost surface, NEXAFS total electron yield spectra (at the M5,4 edges) in the near surface regions, and (N)EXAFS fluorescence spectra (at the L3 edge) in the bulk. Moreover, XRD and EXAFS showed the reversible expansion and contraction of the CeO2 unit cell in H2 and O2 environments, respectively. The expansion of the CeO2 cell was mainly associated with the formation of oxygen vacancies as a result of the Pt-mediated reduction of Ce4+ to Ce3+. We also found that pure mesoporous CeO2 can not be reduced in H2 under identical conditions but can be partially reduced at above 450 °C as revealed by APXPS. The role of Pt in H2 was identified as a catalytic one that reduces the activation barrier for the reduction of CeO2 via hydrogen spillover.
39. Enhanced CO Oxidation Rates at the Interface of Mesoporous Oxides and Pt Nanoparticles
Journal paper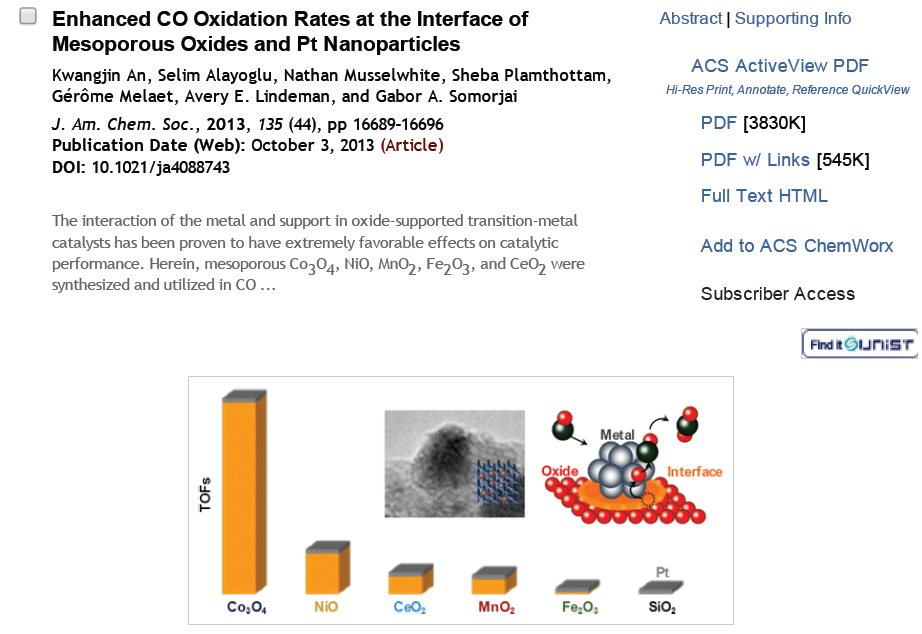
The interaction of the metal and support in oxide-supported transition-metal catalysts has been proven to have extremely favorable effects on catalytic performance. Herein, mesoporous Co3O4, NiO, MnO2, Fe2O3, and CeO2 were synthesized and utilized in CO oxidation reactions to compare the catalytic activities before and after loading of 2.5 nm Pt nanoparticles. Turnover frequencies (TOFs) of pure mesoporous oxides were 0.0002–0.015 s–1, while mesoporous silica was catalytically inactive in CO oxidation. When Pt nanoparticles were loaded onto the oxides, the TOFs of the Pt/metal oxide systems (0.1–500 s–1) were orders of magnitude greater than those of the pure oxides or the silica-supported Pt nanoparticles. The catalytic activities of various Pt/oxide systems were further influenced by varying the ratio of CO and O2 in the reactant gas feed, which provided insight into the mechanism of the observed support effect. In situ characterization using near-edge X-ray absorption fine structure (NEXAFS) and ambient-pressure X-ray photoelectron spectroscopy (APXPS) under catalytically relevant reaction conditions demonstrated a strong correlation between the oxidation state of the oxide support and the catalytic activity at the oxide–metal interface. Through catalytic activity measurements and in situ X-ray spectroscopic probes, CoO, Mn3O4, and CeO2 have been identified as the active surface phases of the oxide at the interface with Pt nanoparticles.
38. Promotion of Hydrogenation of Organic Molecules by Incorporating Iron into Platinum Nanoparticle Catalysts: Displacement of Inactive Reaction Intermediates
Journal paper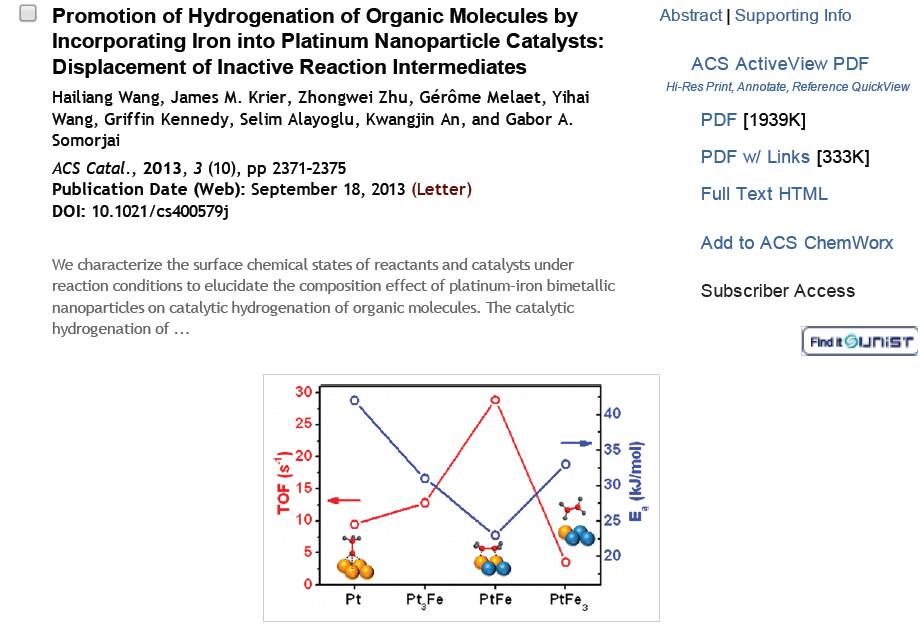
We characterize the surface chemical states of reactants and catalysts under reaction conditions to elucidate the composition effect of platinum−iron bimetallic nanoparticles on catalytic hydrogenation of organic molecules. The catalytic hydrogenation of ethylene is drastically accelerated on the surface of 2 nm PtFe bimetallic nanoparticles as compared to pure Pt. Sum frequency generation (SFG) vibrational spectroscopy indicates that incorporation of Fe into Pt nanoparticle catalysts weakens the adsorption of ethylidyne, an inactive spectator species, on the catalyst surface. Similarly, the turnover frequency of cyclohexene hydrogenation is also significantly enhanced by incorporating Fe into Pt nanoparticle catalysts. Ambient-pressure X-ray photoelectron spectroscopy (AP-XPS) reveals the surface composition and oxidation states of the PtFe nanoparticles under reaction conditions. The oxidation state distribution of Fe responded to the gas atmosphere and the probing depth, whereas the Pt remained largely metallic in all probing conditions. This work represents a molecular level correlation between catalyst structure and catalytic performance.
37. Isomerization of n-Hexane Catalyzed by Supported Monodisperse PtRh Bimetallic Nanoparticles
Journal paperComposition and size of PtxRh1-x bimetallic nanoparticles were varied in order to study the effects in the catalytic reforming of n-hexane. Hexane isomerization, an analogue to the important industrial process of hydrocarbon reforming is a reaction in which we aim to investigate the molecular level details of catalysis. It is known, that in hydrocarbon isomerization, Pt atoms act to isomerize the reactants, while small amounts of “promoter metal” atoms (such as Rh, Ir, Re and Sn) provide C-C and C-H bond breaking activity. Herein, we report on the effect of composition and size in model bimetallic PtxRh1-x nanoparticle catalysts utilized in n-hexane reforming. Both nanoparticle composition and size were shown to influence catalytic turnover frequency and product selectivity. It was found, through ambient pressure X-ray photoelectron spectroscopy, that the surface of these nanoparticles is both dynamic, and Rh rich under relevant reaction conditions. The findings suggest that an ensemble effect exists, in which the highest isomer production occurs when Rh atoms are surrounded by Pt atoms on the metal surface.
36. Influence of Size-Induced Oxidation State of Platinum Nanoparticles on Selectivity and Activity in Catalytic Methanol Oxidation in the Gas Phase
Journal paper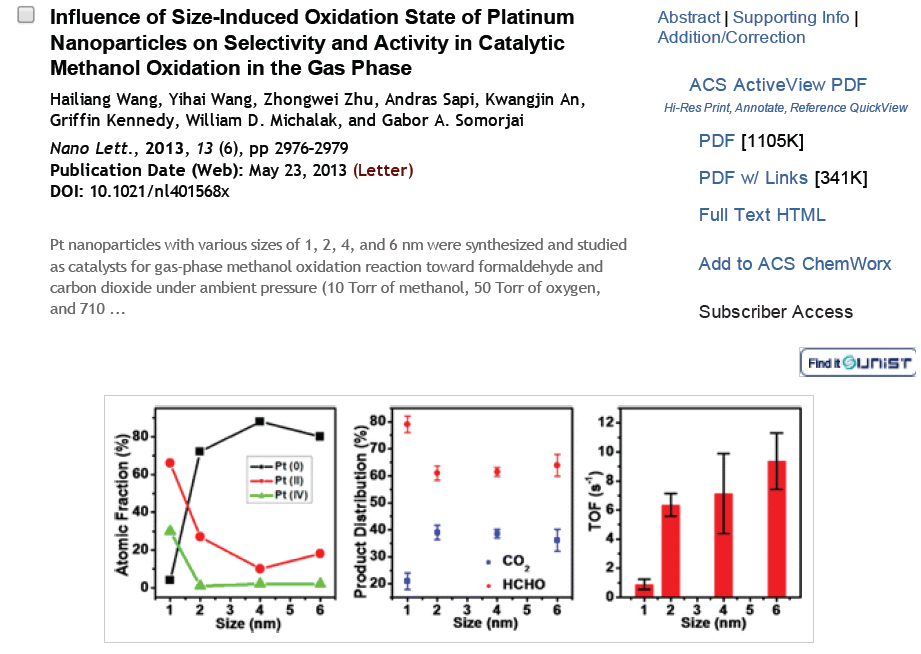
Pt nanoparticles with various sizes of 1, 2, 4, and 6 nm were synthesized and studied as catalysts for gas-phase methanol oxidation reaction toward formaldehyde and carbon dioxide under ambient pressure (10 Torr of methanol, 50 Torr of oxygen, and 710 Torr of helium) at a low temperature of 60 degrees C. While the 2, 4, and 6 rim nanoparticles exhibited similar catalytic activity and selectivity, the 1 nm nanoparticles showed a significantly higher selectivity toward partial oxidation of methanol to formaldehyde, but a lower total turnover frequency. The observed size effect in catalysis was correlated to the size dependent structure and oxidation state of the Pt nanoparticles. X-ray photoelectron spectroscopy and infrared vibrational spectroscopy using adsorbed CO as molecular probes revealed that the 1 nm nanoparticles were predominantly oxidized while the 2, 4, and 6 nm nanoparticles were largely metallic. Transmission electron microscopy imaging witnessed the transition from crystalline to quasicrystalline structure as the size of the Pt nanoparticles was reduced to 1 nm. The results highlighted the important impact of size induced oxidation state of Pt nanoparticles on catalytic selectivity as well as activity in gas-phase methanol oxidation reactions.
35. Preparation of mesoporous oxides and their support effects on Pt nanoparticle catalysts in catalytic hydrogenation of furfural
Journal paper
Mesoporous SiO2, Al2O3, TiO2, Nb2O5, and Ta2O5 were synthesized through a soft-templating approach by a self-assembled framework of Pluronic P123 and utilized for the preparation of 3-dimensional catalysts as supports. Colloidal Pt nanoparticles with an average diameter of 1.9 nm were incorporated into the mesoporous oxides by sonication-induced capillary inclusion. The Pt nanoparticles supported on mesoporous oxides were evaluated in the hydrogenation reaction of furfural (70 tort furfural and 700 torr H2 with a balance of He) to study the effect of catalyst supports on selectivity. In the temperature ranges of 170-240 degrees C, the major products of this reaction were furan, furfuryl alcohol, and 2-methyl furan through a main reaction pathway of either decarbonylation or carbonyl group hydrogenation. While Pt nanoparticles with the size ranges of 1.5-7.1 exhibited strong structure-dependent selectivity, various supports loaded with only 1.9 nm Pt nanoparticles produced dominantly furan as a major product. Compared to the inert silica support, TiO2 and Nb2O5 facilitated an increase in the production of furfuryl alcohol via carbonyl group hydrogenation as a result of a charge transfer interaction between the Pt and the acidic surface of the oxides. The same trend was confirmed on 2-dimensional type catalysts, in which thin films of SiO2, Al2O3, TiO2, Nb2O5, and ZrO2 were prepared as supports. When furfural hydrogenation was conducted (1 torr furfural, 100 torr H2, and 659 torr He) over Pt nanoparticle monolayers deposited on oxide substrates, only TiO2 was shown to increase the production of furfuryl alcohol, while other oxides produced furan.
34. Monodisperse Metal Nanoparticle Catalysts: Synthesis, Characterizations, and Molecular Studies Under Reaction Conditions
Journal paperWe aim to develop novel catalysts that exhibit high activity, selectivity and stability under real catalytic conditions. In the recent decades, the fast development of nanoscience and nanotechnology has allowed synthesis of nanoparticles with well-defined size, shape and composition using colloidal methods. Utilization of mesoporous oxide supports effectively prevents the nanoparticles from aggregating at high temperatures and high pressures. Nanoparticles of less than 2 nm sizes were found to show unique activity and selectivity during reactions, which was due to the special surface electronic structure and atomic arrangements that are present at small particle surfaces. While oxide support materials are employed to stabilize metal nanoparticles under working conditions, the supports are also known to strongly interact with the metals through encapsulation, adsorbate spillover, and charge transfer. These factors change the catalytic performance of the metal catalysts as well as the conductivity of oxides. The employment of new in situ techniques, mainly high-pressure scanning tunneling microscopy (HPSTM) and ambient-pressure X-ray photoelectron spectroscopy (APXPS) allows the determination of the surface structure and chemical states under reaction conditions. HPSTM has identified the importance of both adsorbate mobility to catalytic turnovers and the metal substrate reconstruction driven by gaseous reactants such as CO and O2. APXPS is able to monitor both reacting species at catalyst surfaces and the oxidation state of the catalyst while it is being exposed to gases. The surface composition of bimetallic nanoparticles depends on whether the catalysts are under oxidizing or reducing conditions, which is further correlated with the catalysis by the bimetallic catalytic systems. The product selectivity in multipath reactions correlates with the size and shape of monodisperse metal nanoparticle catalysts in structure sensitive reactions.
33. High Structure Sensitivity of Vapor-Phase Furfural Decarbonylation/Hydrogenation Reaction Network as a Function of Size and Shape of Pt Nanoparticles
Journal paper
Vapor-phase transformations of furfural in H-2 over a series of Pt nanoparticles (NPs) with various particle sizes (1.5-7.1 nm size range) and shapes (rounded, cubes, octahedra) encapsulated in poly(vinylpyrrolidone) (PVP) and dispersed on MCF-17 mesoporous silica were investigated at ambient pressure in the 443-513 K temperature range. Furan and furfuryl alcohol (FFA) were two primary products as a result of furfural decarbonylation and hydrogenation reactions, respectively. Under conditions of the study both reactions exhibited structure sensitivity evidenced by changes in product selectivities, turnover rates (TORs), and apparent activation energies (EA’s) with Pt particle size and shape. For instance, upon an increase in Pt particle size from 1.5 to 7.1 nm, the selectivity toward FFA increases from 1% to 66%, the TOR of FFA production increases from 1 x 10-3 s-1 to 7.6 X 10-2 s-1, and EA decreases from 104 kJ mol-1 to 15 kJ mol-1 (9.3 kPa furfural, 93 kPa H2, 473 K). Conversely, under the same experimental conditions the decarbonylation reaction path is enhanced over smaller nanoparticles. The smallest NPs (1.5 nm) produced the highest selectivity (96%) and highest TOR values (8.8 x 10-2 s-1) toward furan formation. The E-A values for decarbonylation (similar to 62 kJ mol-1) was Pt particle size independent. Furan was further converted to propylene via a decarbonylation reaction, but also to dihydrofuran, tetrahydrofuran, and n-butanol in secondary reactions. Furfuryl alcohol was converted to mostly to 2-methylfuran.
32. Size and Shape Control of Metal Nanoparticles for Reaction Selectivity in Catalysis
Journal paper
A nanoparticle with well-defined surfaces, prepared through colloidal chemistry, enables it to be studied as a model heterogeneous catalyst. The colloidal synthetic approach provides versatile tools to control the size and shape of nanoparticles. Traditional nucleation and growth mechanisms have been utilized to understand how nanoparticles can be uniformly synthesized and unprecedented shapes can be controlled. Now, the size of metal particles can be controlled to cluster regimes by using dendrimers. By using seeds and foreign atoms, specific synthetic environments such as seeded growth and crystal overgrowth can be induced to generate various shaped mono- or bi-metallic, core/shell, or branched nanostructures. For green chemistry, catalysis in 21st century is aiming for 100% selectivity to produce only one desired product at high turnover rates. Recent studies on nanoparticle catalysts clearly demonstrate size and shape dependent selectivity in many catalytic reactions. By combining in situ surface characterization techniques, real-time monitoring of nanoparticles can be performed under reaction environments, thus identifying several molecular factors affecting catalytic activity and selectivity.
31. Sum Frequency Generation Vibrational Spectroscopy of Colloidal Platinum Nanoparticle Catalysts: Disordering versus Removal of Organic Capping
Journal paper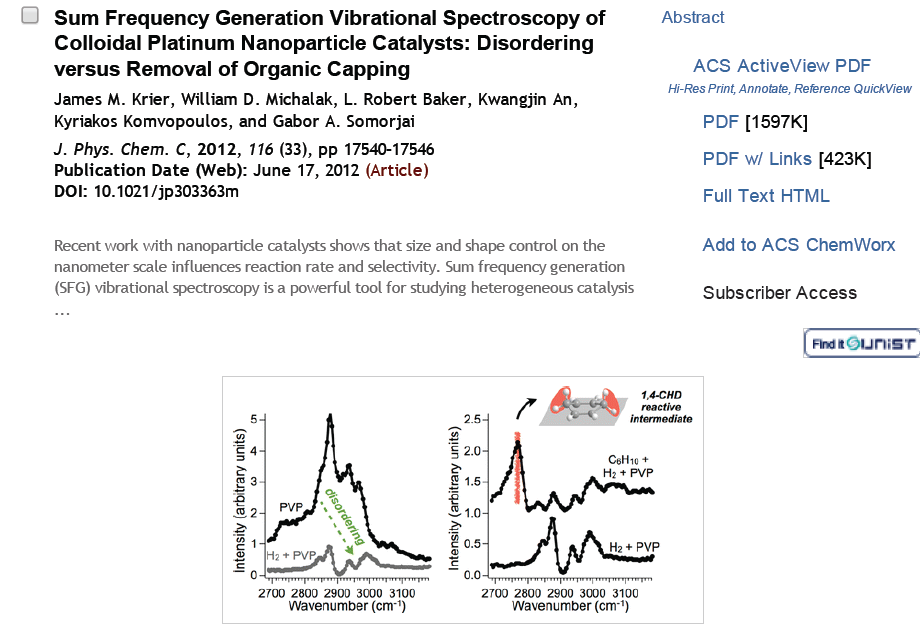
Recent work with nanoparticle catalysts shows that size and shape control on the nanometer scale influences reaction rate and selectivity. Sum frequency generation (SFG) vibrational spectroscopy is a powerful tool for studying heterogeneous catalysis because it enables the observation of surface intermediates during catalytic reactions. To control the size and shape of catalytic nanoparticles, an organic ligand was used as a capping agent to stabilize nanoparticles during synthesis. However, the presence of an organic capping agent presents two major challenges in SFG and catalytic reaction studies: it blocks a significant fraction of active surface sites and produces a strong signal that prevents the detection of reaction intermediates with SFG. Two methods for cleaning Pt nanoparticles capped with poly(vinylpyrrolidone) (PVP) are examined in this study:. solvent cleaning and UV cleaning. Solvent cleaning leaves more PVP intact and relies on disordering with hydrogen gas to reduce the SFG signal of PVP. In contrast, UV cleaning depends on nearly complete removal of PVP to reduce SFG signal. Both UV and solvent cleaning enable the detection of reaction intermediates by SFG. However, solvent cleaning also yields nanoparticles that are stable under reaction conditions, whereas UV cleaning results in aggregation during reaction. The results of this study indicate that solvent cleaning is more advantageous for studying the effects of nanoparticle size and shape on catalytic selectivity by SFG vibrational spectroscopy.
30. Hydrogenation of benzene and toluene over size controlled Pt/SBA-15 catalysts: Elucidation of the Pt particle size effect on reaction kinetics
Journal paper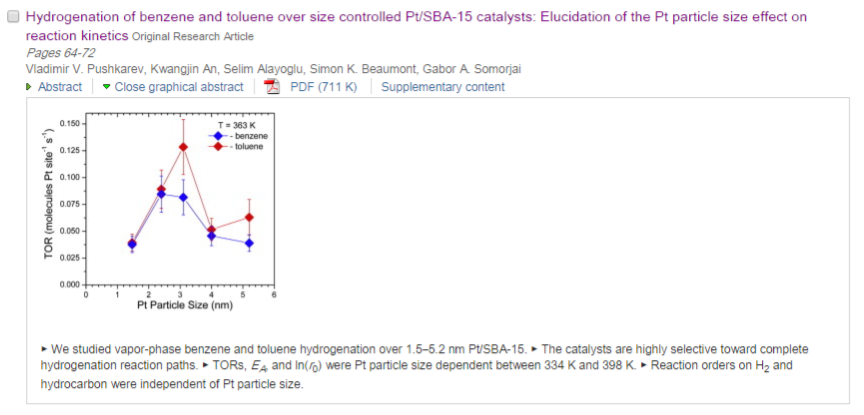
Vapor-phase hydrogenation of benzene and toluene over a size-dependent (1.5-5.2 nm) series of Pt nanoparticles (NPs) encapsulated in polyvinylpyrrolidone (PVP) or polyamidoamine (PAMAM) dendrimer and supported in SBA-15 mesoporous silica was investigated at ambient pressure between 343 K and 398 K. Under these conditions, both reactions exhibit a moderate structure sensitivity evidenced by changes in their turnover rate (TOR), apparent activation energy (EA), pre-exponential factor Inr(o), and deactivation rate parameters with Pt particle size. For instance, the 2.4-3.1 nm Pt NPs showed the highest TOR for both reactions as compared to NPs of either smaller or larger sizes. Also, the E-A and In(ro) factor values were larger for smaller Pt particle sizes, and the rates of catalyst deactivation were more pronounced for catalysts with larger Pt particle sizes. However, the reaction orders on H-2 and hydrocarbon were independent of particle size.
29. Reforming of C-6 Hydrocarbons Over Model Pt Nanoparticle Catalysts
Journal paperSize-controlled model Pt nanoparticle catalysts, synthesized by colloidal chemistry, were used to study the hydrogenative reforming of three C-6 hydrocarbons in mixtures with 5:1 excess of H2: methylcyclopentane, n-hexane and 2-methylpentane. We found a strong particle size dependence on the distribution of different reaction products for the hydrogenolysis of methylcyclopentane. The reactions of 50 Torr methylcyclopentane in 250 Torr H2 at 320 A degrees C, using 1.5 and 3.0 nm Pt nanoparticles produced predominantly C-6 isomers, especially 2-methylpentane, whereas 5.2 and 11.3 nm Pt nanoparticles were more selective for the formation of benzene. For the hydrogenolysis of n-hexane and 2-methylpentane, strong particle size effects on the turnover rates were observed. Hexane and 2-methylpentane reacted up to an order of magnitude slower over 3.0 nm Pt than over the other particle sizes. At 360 A degrees C the isomerization reactions were more selective than the other reaction pathways over 3.0 nm Pt, which also yielded relatively less benzene.
28. Exchange bias behavior of monodisperse Fe3O4/gamma-Fe2O3 core/shell nanoparticles
Journal paperWe have carried out systematic studies on well-characterized monodisperse Fe3O4/gamma-Fe2O3 core/shell nanoparticles of 2-30 nm having a very narrow size distribution and possessing a uniquely mono-layer of surface gamma-Fe2O3. This unique core-shell structure, probably having a disordered magnetic surface state, leads us to three key observations of unusual magnetic properties: i) a very large magnetic exchange anisotropy reaching over 7 x 10(6) erg/cm(3) for the smaller particles, ii) exchange bias behavior in the magnetization data of the core/shell Fe3O4/gamma-Fe2O3 nanoparticles, and iii) the temperature dependence of the coercive field following an unusual exponential behavior.
27. Colloid chemistry of nanocatalysts: A molecular view
Journal paper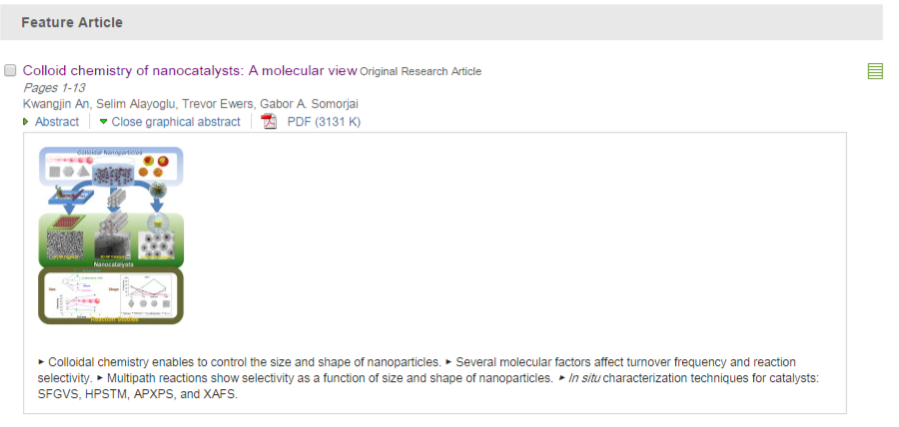
Recent advances of a colloidal chemistry can offer great opportunities to fabricate and design nanocatalysts. Comprehensive understanding of a basic concept and theory of the colloidal synthetic chemistry facilitates to engineer elaborate nano-architectures such as bi- or multi-metallic, heterodimers, and core/shell. This colloidal solution technique not only enables to synthesize high surface mesoporous materials, but also provides a versatile tool to incorporate nanoparticles into mesoporous materials or onto substrates. For green chemistry, catalysis research has been pursued to design and fabricate a catalyst system that produces only one desired product (100% selectivity) at high turnover rates to reduce the production of undesirable wastes. Recent studies have shown that several molecular factors such as the surface structures, composition, and oxidation states affect the turnover frequency and reaction selectivity depending on the size, morphology, and composition of metal nanoparticles. Multipath reactions have been utilized to study the reaction selectivity as a function of size and shape of platinum nanoparticles. In the past, catalysts were evaluated and compared with characterizations before and after catalytic reaction. Much progress on in situ surface characterization techniques has permitted real-time monitoring of working catalysts under various conditions and provides molecular information during the reaction.
26. Synthesis of Uniformly Sized Manganese Oxide Nanocrystals with Various Sizes and Shapes and Characterization of Their T1 Magnetic Resonance Relaxivity
Journal paper
We synthesized manganese oxide (MnO and Mn3O4) nanocrystals with various sizes and shapes by the thermal reaction of a Mn-oleate complex through a heat-up process. When a MnIIoleate complex was thermally decomposed in non-coordinating hydrocarbon solvents, uniformly sized MnO nanocrystals with cubic and octahedral shapes were produced. We were able to synthesize anisotropic, multibranched MnO nanocrystals by the oriented attachment of MnO truncated-nanocube building blocks. When the MnIIoleate complex was heated in 1-hexadecene in the presence of strongly coordinating carboxylic acid surfactants, spherical nanocrystals were generated, and their diameter was controlled in the range 3-13 nm by varying the chain length of the carboxylic acid. When oleyl alcohol was added to the Mnoleate complex in phenyl ether, tetrahedral MnO nanocrystals were synthesized. The as-synthesized MnO nanocrystals were oxidized in air to Mn3O4 or MnO/Mn3O4 coreshell structures, which exhibited exchange coupling with shifted magnetic hysteresis loops. The effect of the size and shape of the phospholipid-capped manganese oxide nanocrystals on their applicability as T1 contrast agents in magnetic resonance imaging (MRI) were examined. As the size of the nanocrystals decreased, their relaxivities increased, thereby generating brighter MR images. In particular, spherical 3 nm-sized Mn3O4 nanocrystals had a high specific relaxivity (r1) of 2.38 mM1s1, clearly demonstrating their potential for use as an efficient T1 MRI contrast agent.
25. Large-Scale Synthesis of Uniform and Extremely Small-Sized Iron Oxide Nanoparticles for High-Resolution T-1 Magnetic Resonance Imaging Contrast Agents
Journal paper
Uniform and extremely small-sized iron oxide nanoparticles (ESIONs) of < 4 nm were synthesized via the thermal decomposition of iron oleate complex in the presence of oleyl alcohol. Oleyl alcohol lowered the reaction temperature by reducing iron oleate complex, resulting in the production of small-sized nanoparticles. XRD pattern of 3 nm-sized nanoparticles revealed maghemite crystal structure. These nanoparticles exhibited very low magnetization derived from the spin-canting effect. The hydrophobic nanoparticles can be easily transformed to water-dispersible and biocompatible nanoparticles by capping with the poly(ethylene glycol)-derivatized phosphine oxide (PO-PEG) ligands. Toxic response was not observed with Fe concentration up to 100 mu g/mL in MTT cell proliferation assay of POPEG-capped 3 nm-sized iron oxide nanoparticles. The 3 nm-sized nanoparticles exhibited a high r1 relaxivity of 4.78 mM-1s-1 and low r2/r1 ratio of 6.12, demonstrating that ESIONs can be efficient T-1 contrast agents. The high r1 relaxivities of ESIONs can be attributed to the large number of surface Fe3+ ions with 5 unpaired valence electrons. In the in vivo T-1-weighted magnetic resonance imaging (MRI), ESIONs showed longer circulation time than the clinically used gadolinium complex-based contrast agent, enabling high-resolution imaging. High-resolution blood pool MR imaging using ESIONs enabled clear observation of various blood vessels with sizes down to 0.2 mm. These results demonstrate the potential of ESIONs as T-1 MRI contrast agents in clinical settings.
24. Large-Scale Synthesis of Ultrathin Manganese Oxide Nanoplates and Their Applications to T1 MRI Contrast Agents
Journal paper
Lamellar structured ultrathin manganese oxide nanoplates have been synthesized from thermal decomposition of manganese(II) acetylacetonate in the presence of 2,3-dihydroxynaphthalene, which promoted two-dimensional (2-D) growth by acting not only as a strongly binding surfactant but also as a structure-directing agent. Ultrathin manganese oxide nanoplates with a thickness of about 1 rim were assembled into a lamellar structure, and the width of the nanoplates could be controlled from 8 to 70 nm by using various coordinating solvents. X-ray absorption near-edge structure (XANES) spectra at the Mn K edge clearly showed that the nanoplates are mainly composed of Mn(II) species with octahedral symmetry. These hydrophobic manganese oxide nanoplates were ligand-exchanged with amine-terminated poly(ethyleneglycol) to generate water-dispersible nanoplates and applied to T1 contrast agents for magnetic resonance imaging (MRI). They exhibited a very high longitudinal relaxivity (r1) value of up to 5.5 mM-1s-1 derived from their high concentration of manganese ions exposed on the surface, and strong contrast enhancement of in vitro and in vivo MR images was observed with a very low dose.
23. In vitro cytotoxicity screening of water-dispersible metal oxide nanoparticles in human cell lines
Journal paperIn this study, we present in vitro cytotoxicity of iron oxide (Fe3O4) and manganese oxide (MnO) using live/dead cell assay, lactate dehydrogenase assay, and reactive oxygen species detection with variation of the concentration of nanoparticles (5-500 mu g/ml), incubation time (18-96 h), and different human cell lines (lung adenocarcinoma, breast cancer cells, and glioblastoma cells). The surface of nanoparticles is modified with polyethyleneglycol-derivatized phospholipid to enhance the biocompatibility, water-solubility, and stability under an aqueous media. While the cytotoxic effect was negligible for 18 h incubation even at highest concentration of 500 mu g/ml, MnO nanoparticle represented higher level of toxicity than those of Fe3O4 and the commercial medical contrast reagent, Feridex after 2 and 4 day incubation time. However, the cytotoxicity of Fe3O4 is equivalent or better than Feridex based on the live/dead cell viability assay. The engineered MnO and Fe3O4 exhibited excellent stability compared with Feridex for a prolonged incubation time.
22. Synthesis and biomedical applications of hollow nanostructures
Journal paperHollow nanostructures have attracted tremendous attention from researchers in various disciplines because their high surface to volume ratio and large pore volume are highly desirable for many technological applications including drug delivery system. Several colloidal synthetic methods have been used to synthesize various hollow nanostructures. These synthetic approaches are mainly categorized into four main classes according to how the hollow structure is formed: the Kirkendall effect, chemical etching, galvanic replacement, and template-mediated approach. The large pores inside the hollow nanostructures can encapsulate and release various drugs and biomolecules, while the surface of the nanostructure can be functionalized for drug targeting or bio-labeling. These features make the hollow nanostructures a unique and promising candidate as multifunctional drug delivery vehicles. This review article covers recent progress concerning the synthesis of hollow nanostructures with their sizes smaller than 200 nm and their biomedical, applications including specific targeting, imaging, and controlled release of therapeutics for simultaneous diagnosis and therapy.
21. Simple and Generalized Synthesis of Semiconducting Metal Sulfide Nanocrystals
Journal paper
Uniform-sized semiconducting nanocrystals of binary metal sulfides are synthesized from the thermolysis of metal-oleate complexes in alkanethiol. The size of the Cu2S nanocrystals can be tuned from 7 to 20 nm by varying the reaction conditions. Various shaped nanocrystals of CdS, ZnS, MnS, and Pb are synthesized from the thermal reaction of metal-oleate in alkanethiol.
20. Synthesis of Uniform Ferrimagnetic Magnetite Nanocubes
Journal paper
We synthesized uniform ferrimagnetic magnetite nanocubes in the size range from 20 to 160 nm. The magnetic property of the nanocubes was characterized, and magnetic separation of the histidine-tagged protein was demonstrated.
19. Large-Scale Soft Colloidal Template Synthesis of 1.4 nm Thick CdSe Nanosheets
Journal paper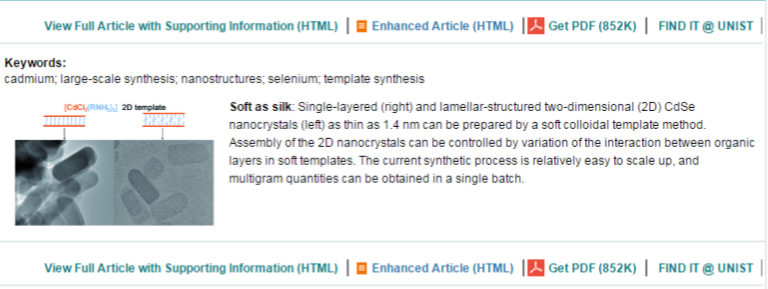
Single-layered (right) and lamellar-structured two-dimensional (2D) CdSe nanocrystals (left) as thin as 1.4 nm can be prepared by a soft colloidal template method. Assembly of the 2D nanocrystals can be controlled by variation of the interaction between organic layers in soft templates. The current synthetic process is relatively easy to scale up, and multigram quantities can be obtained in a single batch.
18. Synthesis of Uniform Hollow Oxide Nanoparticles through Nanoscale Acid Etching
Journal paper
We synthesized various hollow oxide nanoparticles from as-prepared MnO and iron oxide nanocrystals. Heating metal oxide nanocrystals dispersed in technical grade trioctylphosphine oxide (TOPO) at 300 degrees C for hours yielded hollow nanoparticles retaining the size and shape uniformity of the original nanocrystals. The method was highly reproducible and could be generalized to synthesize hollow oxide nanoparticles of various sizes, shapes, and compositions. Control experiments revealed that the impurities in technical grade TOPO, especially alkylphosphonic acid, were responsible for the etching of metal oxide nanocrystals to the hollow structures. Elemental mapping analysis revealed that the inward diffusion of phosphorus and the outward diffusion of metal took place in the intermediate stages during the etching process. The elemental analysis using XPS, EELS, and EDX showed that the hollow nanoparticles were amorphous metal oxides containing significant amount of phosphorus. The hollow nanoparticles synthesized from MnO and iron oxide nanocrystals were paramagnetic at room temperature and when dispersed in water showed spin relaxation enhancement effect for magnetic resonance imaging (MRI). Because of their morphology and magnetic property, the hollow nanoparticles would be utilized for multifunctional biomedical applications such as the drug delivery vehicles and the MRI contrast agents.
17. Simple and Generalized Synthesis of Oxide-Metal Heterostructured Nanoparticles and their Applications in Multimodal Biomedical Probes
Journal paper
Heterostructured nanoparticles composed of metals and Fe3O4 or MnO were synthesized by thermal decomposition of mixtures of metal-oleate complexes (for the oxide component) and metal-oleylamine complexes (for the metal component). The products included flowerlike-shaped nanoparticles of Pt- Fe3O4 and Ni- Fe3O4 and snowmanlike-shaped nanoparticles of Ag-MnO and Au-MnO. Powder X-ray diffraction patterns showed that these nanoparticles were composed of face-centered cubic (fcc)-structured Fe3O4 or MnO and fcc-structured metals. The relaxivity values of the Au-MnO and Au- Fe3O4 nanoparticles were similar to those of the MnO and Fe3O4 nanoparticles, respectively. Au-Fe3O4 heterostructured nanoparticles conjugated with two kinds of 12-base oligonucleotide sequences were able to sense a complementary 24-mer sequence, causing nanoparticle aggregation. This hybridization-mediated aggregation was detected by the overall size increase indicated by dynamic light scattering data, the red shift of the surface plasmon band of the Au component, and the enhancement of the signal intensity of the Fe3O4 component in T(2)-weighted magnetic resonance imaging.
16. Monodispersed MnO nanoparticles with epitaxial Mn3O4 shells
Journal paperWe report the microstructural and magnetic properties of monodispersed nanoparticles (NPs) of antiferromagnetic MnO ( T-N = 118 K), with epitaxial ferrimagnetic Mn3O4 (T-C = 43 K) shells. Above T-C, an unusually large magnetization is present, produced by the uncompensated spins (UCSs) on the surface of the MnO particles. These spins impart a net anisotropy to the MnO particles that is approximately three orders of magnitude larger than the bulk value. As a result, an anomalously high blocking temperature is exhibited by the MnO particles, and finite coercivity and exchange bias are present above T-C. When field cooled below T-C, a strong exchange bias was established in the Mn3O4 shells as a result of high net anisotropy of the MnO particles. A large coercivity was also observed. Models of several aspects of the behaviour of this unusual system emphasized the essential role of the UCSs on the surfaces of the MnO NPs.
Rat glioma cells were labeled using electroporation with either manganese oxide (MnO) or superparamagnetic iron oxide (SPIO) nanoparticles. The viability and proliferation of SPIO-labeled cells (1.9 mg Fe/ml) or cells electroporated with a low dose of MnO (100 mu g Mn/ml) was not significantly different from unlabeled cells; a higher MnO dose (785 mu g Mn/ml) was found to be toxic. The cellular ion content was 0.1-0.3 pg Mn/cell and 4.4 pg Fe/cell, respectively, with cellular relaxivities of 2.5-4.8 s-1 (RI) and 45-84 s-1 (R2) for MnO-labeled cells. Labeled cells (SPIO and low-dose MnO) were each transplanted in contralateral brain hemispheres of rats and imaged in vivo at 9.4T. While SPIO-labeled cells produced a strong “negative contrast” due to the increase in R2, MnO-Iabeled cells produced “positive contrast” with an increased R1. Simultaneous imaging of both transplants with opposite contrast offers a method for MR “double labeling” of different cell populations.
14. Antiferromagnetic MnO nanoparticles with ferrimagnetic Mn3O4 shells: Doubly inverted core-shell system
Journal paperWe report the magnetic and microstructural properties of antiferromagnetic MnO nanoparticles with shells of ferrimagnetic Mn3O4which is opposite the usual arrangement of antiferromagnetically coated ferromagnetic nanoparticles. In addition, the antiferromagnetic MnO cores order at much higher temperature (TN=118 K) than the ferrimagnetic Mn3O4 shells (TC=43 K)-another reversal of the usual situation. The single crystal MnO cores, with rocksalt structure, are crystallographically aligned with the tetragonal spinel structure of the Mn3O4 shells. Particles field cooled in 50 kOe have large coercive force and exchange bias below TC, e.g., 5800 and 2950 Oe, respectively, at 5 K. The spontaneous magnetization at TC( Mn3O4)is similar to 20% of its value at 5 K, and remains finite for more than 20 K above TC( Mn3O4). Hysteresis with exchange bias is present in this anomalous region. The MnO cores with their uncompensated spins are responsible for the behavior above TC(Mn3O4). The MnO cores have a blocking temperature of 95 K, and the hysteresis and exchange bias above TC(Mn3O4) results from the switching of the MnO spin lattices by their uncompensated spins. Analysis of the thermoremanent magnetization and field cooling and/or zero field cooling in 50 kOe, and the dependence of exchange bias on the temperature at which the cooling field was applied support this model.
13. Synthesis of uniform goethite nanotubes with parallelogram cross section
Journal paperUniform goethite nanotubes were synthesized from the reaction of hydrazine with Fe(III)-oleate complex immobilized in reverse micelles. The nanotubes have interesting parallelogram cross section with uniform edge dimension of as small as 7 nm. The edge dimensions and lengths of the nanotubes were easily controlled by varying the reaction conditions.
12. 57Fe Mossbauer spectral and muon spin relaxation study of the magnetodynamics of monodispersed gamma-Fe2O3 nanoparticles
Journal paperThe Mossbauer spectra of monodispersed iron oxide nanoparticles with diameters of 4, 7, 9, and 11 nm have been measured between 4.2 and 315 K and fitted within the formalism for stochastic fluctuations of the hyperfine Hamiltonian. In this model, the hyperfine field is assumed to relax between the six +/- x, +/- y, and +/- z directions in space with a distribution of relaxation rates that is temperature dependent. Muon spin relaxation measurements have been carried out on the 9 nm particles between 4.2 and 295 K. Both techniques reveal three regimes in the magnetic dynamics of these nanoparticles. In the low-temperature regime, between 4.2 and similar to 30 K, the nanoparticle magnetic moments are blocked and a spin-glass-like state is observed with nearly static hyperfine fields, as is indicated by the well resolved magnetic Mossbauer spectra and the slow exponential decay of the muon asymmetry functions. In the high-temperature regime, above similar to 125 K, the nanoparticle magnetic moments and, hence, the hyperfine fields, relax rapidly and a typical thermally activated superparamagnetic behavior is observed, as is indicated by the Mossbauer doublet line shape and the muon asymmetry functions that are unquestionably characteristic of monodispersed nanoparticles. In the intermediate regime between similar to 30 and 125 K, the Mossbauer spectra are the superposition of broad sextets and doublets and the muon asymmetry functions have been fitted with a sum of two terms, one relaxing term similar to that observed at and above 125 K and one term characteristic of static local fields. Hence, in this intermediate regime, the sample is magnetically inhomogeneous and composed of nanoparticles rapidly and slowly relaxing as a result of interparticle interactions. The magnetic anisotropy constants determined from both the Mossbauer spectral and magnetic susceptibility results decrease by a factor similar to 4 with increasing diameter from 4 to 22 nm and increase linearly with the percentage of iron(III) ions present at the surface of the nanoparticles. The interparticle interaction energy is estimated to be between 89 and 212 K from the temperature dependence of the magnetic hyperfine field measured on the 9 nm nanoparticles.
11. Experimental studies of strong dipolar interparticle interaction in monodisperse Fe3O4 nanoparticles
Journal paperInterparticle interaction of monodisperse Fe3O4 nanoparticles has been experimentally investigated by dispersing the nanoparticles in solvents. With increasing the interparticle distances to larger than 100 nm in a controlled manner, the authors found that the blocking temperature (T-B) of the nanoparticles drops continuously and eventually gets saturated with a total drop in TB of 7-17 K observed for 3, 5, and 7 nm samples, compared with their respective nanopowder samples. By carefully studying the dependence of TB on the interparticle distance, the authors could demonstrate that the experimental dependence of T-B follows the theoretical curve of the dipole-dipole interaction.

Hollow Fe nanoframes were synthesized from the thermal decomposition of Fe(II)-stearate complex in the presence of sodium oleate and oleic acid. Sodium molten salts derived from sodium oleate seemed to be responsible for the formation of the nanoframes.
9. Development of a T-1 contrast agent for magnetic resonance imaging using MnO nanoparticles
Journal paper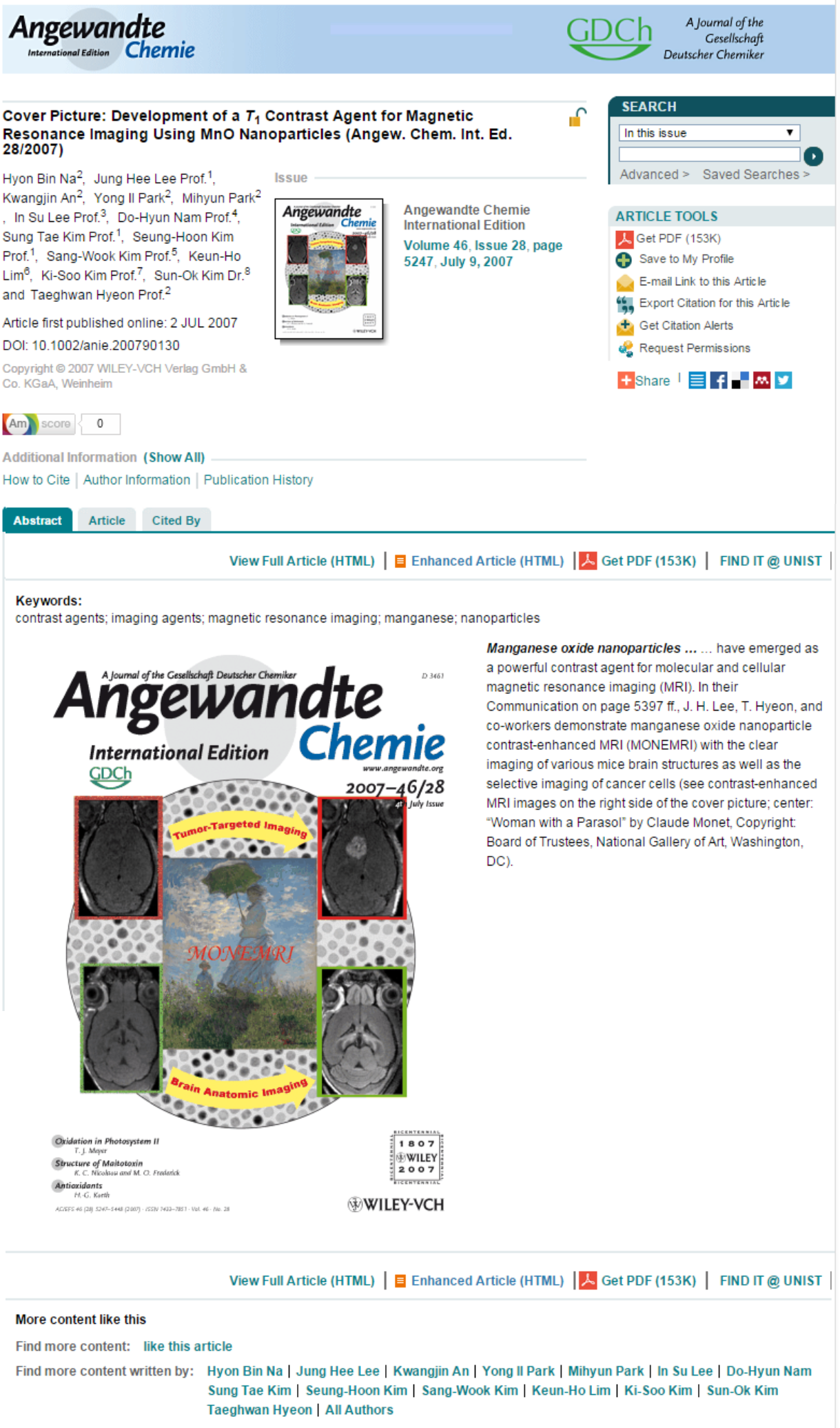
A new T1 contrast agent for magnetic resonance imaging (MRI) based on MnO nanoparticles reveals a bright signal enhancement and fine anatomic structures in the T1-weighted MR image of a mouse brain (see picture; left MRI, right MnO-enhanced MRI (MONEMRI)). Furthermore, MnO nanoparticles conjugated with a tumor-specific antibody were used for selectively imaging breast cancer cells in a metastatic tumor in brain.
8. Inter-particle and interfacial interaction of magnetic nanoparticles
Journal paperIn order to understand inter-particle as well as interfacial interaction of magnetic nanoparticles, we have prepared several Fe3O4 nanoparticles in the ranges from 3 to 50 nm. These nanoparticles are particularly well characterized in terms of size distribution with a standard deviation (sigma) in size less than 0.4 nm. We investigated the inter-particle interaction by measuring the magnetic properties of the nanoparticles while controlling inter-particle distances by diluting the samples with solvents. According to this study, blocking temperatures dropped by 8-17 K with increasing the inter-particle distances from a few nm to 140 nm while the overall shape and qualitative behavior of the magnetization remain unchanged. It implies that most features observed in the magnetic properties of the nanoparticles are due to the intrinsic properties of the nanoparticles, not due to the inter-particle interaction. We then examined possible interfacial magnetic interaction in the core-shell structure of our Fe3O4 nanoparticles.
7. Synthesis, characterization, and self-assembly of pencil-shaped CoO nanorods
Journal paper
We synthesized uniformly sized, pencil-shaped CoO nanorods by the thermal decomposition of a cobalt-oleate complex, which was prepared from the reaction of cobalt chloride and sodium oleate. The diameters and lengths of the CoO nanorods were easily controlled by varying the experimental conditions, such as the heating rate and the amount of Co-oleate complex. The X-ray diffraction pattern revealed that the CoO nanorods have an extraordinary wurtzite ZnO crystal structure. These uniformly sized nanorods self-assembled to form both horizontal parallel arrangements and perpendicular hexagonal honeycomb superlattice structures. Reduction of the nanorods by heating under a hydrogen atmosphere generated either hcp Co or Co2C nanorods. Characterization of the CoO nanorods using X-ray absorption spectroscopy, X-ray magnetic circular dichroism spectroscopy, and magnetic measurements showed that they contain a small fraction of ferromagnetic Co impurities.
4. Magnetic fluorescent delivery vehicle using uniform mesoporous silica spheres embedded with monodisperse magnetic and semiconductor nanocrystals
Journal paper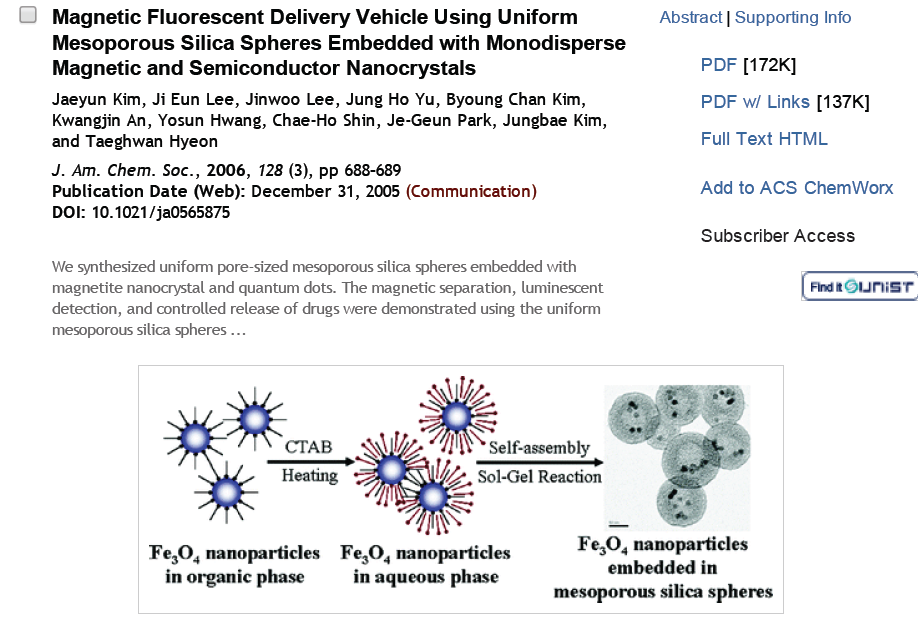
We synthesized uniform pore-sized mesoporous silica spheres embedded with magnetite nanocrystal and quantum dots. The magnetic separation, luminescent detection, and controlled release of drugs were demonstrated using the uniform mesoporous silica spheres embedded with monodisperse nanocrystals.
5. Sea urchin shaped carbon nanostructured materials: carbon nanotubes immobilized on hollow carbon spheres
Journal paper
Novel sea urchin shaped nanostructured carbon spheres (carbon nano-urchins) were fabricated by the growth of carbon nanotubes on the surface of hollow carbon spheres. The carbon nano-urchins were successfully used as a catalyst support for methanol electrochemical oxidation.
6. Generalized fabrication of multifunctional nanoparticle assemblies on silica spheres
Journal paper97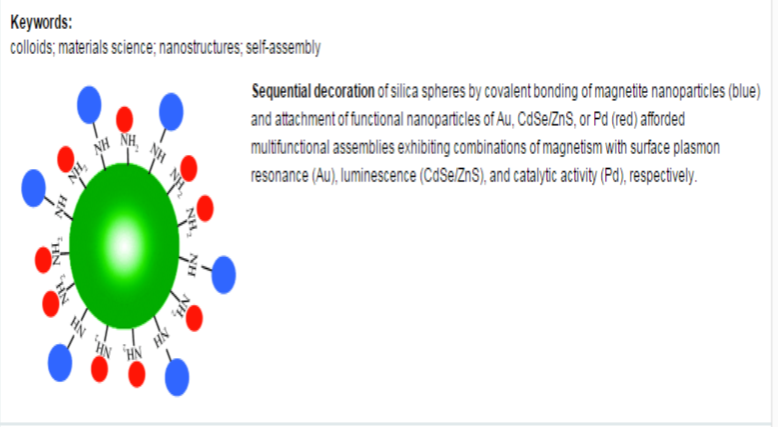
Sequential decoration of silica spheres by covalent bonding of magnetite nanoparticles (blue) and attachment of functional nanoparticles of Au, CdSe/ZnS, or Pd (red) afforded multifunctional assemblies exhibiting combinations of magnetism with surface plasmon resonance (Au), luminescence (CdSe/ZnS), and catalytic activity (Pd), respectively.
3. Large-scale synthesis of hexagonal pyramid-shaped ZnO nanocrystals from thermolysis of Zn-oleate complex
Journal paper
We report the large-scale synthesis of uniform-sized hexagonal pyramid-shaped ZnO nanocrystals by the thermolysis of Zn-oleate complex, which was prepared from the reaction of inexpensive and environmentally friendly reagents such as zinc chloride and sodium oleate. Under optimized reaction conditions, we were able to synthesize as much as 2.83 g using 300 mL of oleylamine and 90 mL (284 mmol) of oleic acid (OA) as both solvents and stabilizing surfactants. The UV-vis spectrum showed the absorption onset of 380 nm, and the photoluminescence spectrum showed a near band-edge emission at 387 nm and a broad blue-green emission band above 468 nm.
2. Ultra-large-scale syntheses of monodisperse nanocrystals
Journal paper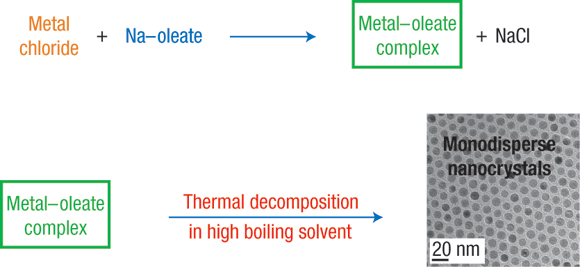
The development of nanocrystals has been intensively pursued, not only for their fundamental scientific interest, but also for many technological applications. The synthesis of monodisperse nanocrystals (size variation <5%) is of key importance, because the properties of these nanocrystals depend strongly on their dimensions. For example, the colour sharpness of semiconductor nanocrystal-based optical devices is strongly dependent on the uniformity of the nanocrystals and monodisperse magnetic nanocrystals are critical for the next-generation multi-terabit magnetic storage media. For these monodisperse nanocrystals to be used, an economical mass-production method needs to be developed. Unfortunately, however, in most syntheses reported so far, only sub-gram quantities of monodisperse nanocrystals were produced. Uniform-sized nanocrystals of CdSe and Au have been produced using colloidal chemical synthetic procedures. In addition, monodisperse magnetic nanocrystals such as Fe, Co, gamma-Fe2O3, and Fe3O4 have been synthesized by using various synthetic methods23. Here, we report on the ultra-large-scale synthesis of monodisperse nanocrystals using inexpensive and non-toxic metal salts as reactants. We were able to synthesize as much as 40 g of monodisperse nanocrystals in a single reaction, without a size-sorting process. Moreover, the particle size could be controlled simply by varying the experimental conditions. The current synthetic procedure is very general and nanocrystals of many transition metal oxides were successfully synthesized using a very similar procedure.
1. Novel synthesis of magnetic Fe2P nanorods from thermal decomposition of continuously delivered precursors using a syringe pump
Journal paper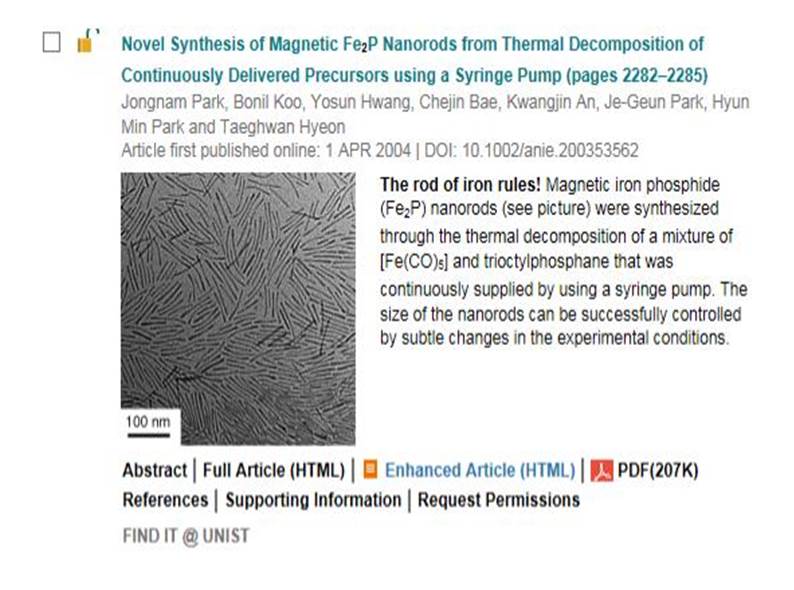
Magnetic iron phosphide (Fe2P) nanorods (see picture) were synthesized through the thermal decomposition of a mixture of [Fe(CO)5] and trioctylphosphane that was continuously supplied by using a syringe pump. The size of the nanorods can be successfully controlled by subtle changes in the experimental conditions.